Texto universitario

_____________________________
Módulo 4. Agua
4.1 Joseph Priestley y la revolución química
A mediados del siglo XVIII la gente todavía consideraba el agua como un elemento. Para los europeos, esta idea se remonta al menos a los antiguos griegos de la época de Aristóteles, según los cuales el agua era uno de los cuatro elementos básicos (junto con la tierra, el aire y el fuego) que constituían todas las sustancias que componían el mundo terrestre. Tales incluso había postulado que el agua era el elemento del que estaba hecho todo. Ahora sabemos que el agua no es un elemento, sino un compuesto formado por oxígeno e hidrógeno. Aquí contamos la historia de cómo llegamos a saber eso, como resultado de la Revolución Química hace poco más de 200 años. Es una historia que se ha contado muchas veces, y los lectores expertos pueden preguntarse por qué debería intentar contarla de nuevo. La razón es simple: con demasiada frecuencia se dice todo mal, “incorrecto” en varios sentidos: incorrecto acerca de las circunstancias históricas, ignorante de los argumentos científicos relevantes, crítico sobre la base de profundos malentendidos y filosóficamente ingenuo y simplista. Los mejores conocimientos disponibles suelen estar enterrados en trabajos especializados que son ignorados incluso por la mayoría de historiadores profesionales y filósofos de la ciencia.
Esperemos que al contar esta historia aumente su interés en la Revolución Química como un tema apasionante del pensamiento histórico, filosófico y científico.
Joseph Priestley
Nuestra historia comienza con Joseph Priestley (1733–1804), un típico personaje de la ciencia aficionada del siglo XVIII[1]. Predicador disidente y consultor político, Priestley floreció en la benigna exclusión impuesta por la ortodoxia anglicana. Era uno de esos hombres de ciencia británicos que nunca se acercaba a una universidad, ni para aprender ni para enseñar. Su investigación científica se llevó a cabo en casa, inicialmente en las “cocinas de las cabañas de Yorkshire con micropisos cálidos[2]”. Su gran trabajo en química comenzó cuando se mudó a Leeds en 1767, donde tuvo la suerte de “habitar una casa contigua a una cervecería pública[3]”. Experimentando con el “aire fijo” que se encontraba acumulado en las tinas de fermentación (lo que ahora llamamos dióxido de carbono), se convirtió en una celebridad en toda Europa cuando encontró una forma de hacer agua artificialmente carbonatada. Este trabajo también marcó el comienzo de su programa de investigación a largo plazo en "química neumática", la química de los gases, o "aires", como él y sus contemporáneos los concibieron más comúnmente.
Priestley fue el más prolífico descubridor y fabricante de nuevos aires. No mucho antes de su trabajo, la mayoría de la gente había considerado el aire como un elemento puro tanto como el agua, aunque antes se habían informado algunas observaciones aisladas de ciertos gases diferentes del aire ordinario. Después de su trabajo, no quedó ninguna duda de que el aire ordinario tenía al menos dos componentes y que se podían producir diferentes tipos de aire mediante diversas reacciones químicas. Priestley's Experiments and Observations on Different Kinds of Air (1774, 1790) es un puro deleite para aquellos que comparten un sentido de fascinación por todos los diversos fenómenos de la naturaleza y un asombro infantil por nuestra propia capacidad para provocarlos. Priestley fue la primera persona en hacer y embotellar lo que ahora llamamos oxígeno y contarlo al mundo entero. En la divertida obra de Carl Djerassi y Roald Hoffmann, Oxígeno (2001), no se sabe quién debería ganar el primer "Premio Retro-Nobel" de Química por el descubrimiento del oxígeno[4]. Pero esos autores, o cualquier otra persona suficientemente informada, no negarían la prioridad de Priestley sobre Carl Wilhelm Scheele (1742-1786) en la publicación y sobre Antoine-Laurent Lavoisier (1743-1794) en los hechos. El entusiasmo de Priestley es palpable cuando informa, en su carta del 15 de marzo de 1775 a James Pringle, presidente de la Royal Society de Londres: “el más notable de todos los tipos de aire que he producido. . . es uno que es cinco o seis veces mejor que el aire común, a los efectos de la respiración, la inflamación y, creo, cualquier otro uso del aire atmosférico común". Primero probó este nuevo aire quemando cosas en él. Y luego, “para completar la prueba de la calidad superior de este aire, le introduje un ratón; y en una cantidad en la que, de haber estado en el aire común, habría muerto en aproximadamente un cuarto de hora, vivió, en dos momentos diferentes, una hora entera, y fue sacado con bastante vigor”. Después de eso, encontró el valor para respirar el aire nuevo él mismo. “La sensación en mis pulmones”, informó Priestley, “no era sensiblemente diferente a la del aire común, pero imaginé que mi pecho se sentía particularmente ligero y suave durante algún tiempo después. Quién sabe si, con el tiempo, este aire puro puede convertirse en un artículo de moda en el lujo. Hasta ahora, solo dos ratones y yo hemos tenido el privilegio de respirarlo".
En el nuevo sitio de la Capilla Mill Hill en Leeds, por el cual predicó durante varios años durante su apogeo científico, se le cita: “Joseph Priestley, descubridor del oxígeno, fue ministro aquí 1767-1773”. Tal conmemoración habría molestado a Priestley, porque no llamó a su nuevo gas "oxígeno". Lo llamó “aire desflogistizado”, y eso no fue solo una cuestión de palabras. Con esa frase realmente se refería al aire común limpio del "flogisto" que normalmente se mezcla en él. ¿Qué era el flogisto? En resumen, era el principio de inflamabilidad; “Principio” aquí no significaba una regla fundamental, sino una sustancia fundamental que se combinaba con otras sustancias y les daba sus propiedades características. El flogisto fue el principio que impartió combustibilidad a los combustibles. Una sustancia combustible era rica en flogisto, y al arder liberaba su flogisto, que luego se manifestaba en la llama que salía.
Ciertos experimentos parecían indicar que los metales también eran ricos en flogisto, y que era el flogisto el que les daba las propiedades metálicas características, como su brillo brillante, su maleabilidad y ductilidad y su conductividad eléctrica (y su inflamabilidad en realidad, en las circunstancias adecuadas). Cuando un metal fue privado de flogisto, perdió sus propiedades metálicas clave y se convirtió en una sustancia terrosa llamada "calx" (que ahora identificaríamos como herrumbre u óxido). Todo esto suena demasiado fantasioso para nuestro oído moderno. Veamos si podemos hacer que el flogisto-teórico vea algo de sentido común. Si la cal es realmente un metal que ha perdido su flogisto, entonces deberías poder convertirlo de nuevo en metal dándole un poco de flogisto. ¿Puedes hacer eso? “Claro”, dice el flogisto. Eso es lo que han estado haciendo las fundiciones durante miles de años. Tome un mineral metálico, que a menudo contiene cal en lugar de metal puro, y mézclelo con una sustancia rica en flogisto, por ejemplo, carbón vegetal; Caliente la mezcla fuertemente, para efectuar una transferencia de flogisto del carbón vegetal al calx. ¡Y ahí viene, el metal brillante! Un trabajo similar sobre la interconversión de azufre y ácido sulfúrico realizado por el médico y químico alemán Georg Ernst Stahl (1659-1734) fue uno de los experimentos fundacionales de la química flogistonista[5]. Este trabajo despertó la admiración de Immanuel Kant, quien lo eligió como una de las tres ilustraciones principales de cómo la ciencia empírica comenzó a lidiar con la naturaleza de una manera basada en principios, en su Crítica de la razón pura: cuando “Stahl transformó metales en calx este último de vuelta al metal quitando primero algo y luego volviéndolo a colocar, una luz se alzó para todos aquellos que estudian la naturaleza[6] ".
De manera similar, Priestley había producido su oxígeno (aire desflogistizado) a través de un proceso en el que pensaba que la cal de mercurio se “revivía” en su forma metálica al absorber el flogisto del aire. Como resultado, el aire habría sido "desflogistizado". Un aire así debería ser un soporte excepcionalmente bueno de la combustión, ya que reabsorbería el flogisto con mucho entusiasmo. Y así fue cuando Priestley intentó el experimento.
También razonó que, dado que la respiración era un proceso en el que el flogisto producido por el funcionamiento del cuerpo se eliminaba de los pulmones, el aire desflogistizado debería ser particularmente bueno para respirar. Y así fue. Uno puede ver por qué Georges Cuvier bromeó diciendo que Priestley era un padre de la química moderna, pero "un padre que nunca quiso reconocer a su hija[7]". Priestley es a menudo visto como una figura trágica, poseedor de una habilidad experimental consumada y lleno de buenas intenciones científicas, pero cegado por una adherencia dogmática a una forma de pensar anticuada. Su desgracia química se vio agravada por la injusticia política, cuando una turba reaccionaria saqueó su casa y laboratorio en Birmingham en 1791 por su apoyo a la Revolución Francesa, en el segundo aniversario del asalto a la Bastilla. Después de eso, intentó una vida en Londres, pero al final solo encontró refugio en Estados Unidos. Se dice que fue triste, pero inevitablemente correcto, que su obra fuera barrida por la marea del progreso científico que trajo Antoine-Laurent Lavoisier, el joven parisino urbano con brillantez y ambición en igual medida. Lavoisier tenía una forma diferente de explicar los experimentos y observaciones de Priestley. La combustión se combinó con oxígeno, al igual que la calcinación (convertir el metal en cal). Donde Priestley vio la desflogisticación, Lavoisier vio la oxidación. Habiendo visto la luz mostrada por Lavoisier, los químicos nunca han mirado atrás al flogisto… Incluso Thomas Kuhn, quien se negó a decir que el lado perdedor de una revolución científica estaba simplemente equivocado, estaba sorprendentemente deprimido con Priestley. Aunque negó que la resistencia de Priestley a la química lavoisieriana fuera alguna vez "ilógica o poco científica", Kuhn pensó que era "irrazonable" resistir tanto tiempo como lo hizo; el historiador “tal vez desee decir que el hombre que continúa resistiendo después de que toda su profesión se ha convertido ha dejado ipso facto de ser científico[8]”. La historia del viejo y terco Priestley cegado por el dogma flogístico ha capturado la imaginación de muchas personas, pero es una historia engañosa en muchos niveles. La mejor manera de empezar a ver el problema es preguntarse: ¿qué estaba tan mal en la postura de Priestley?
Hasta el final de su vida continuó su investigación química y publicó defensas del flogisto bien informadas y rigurosamente razonadas. Continuó encontrando la teoría del flogisto como un marco sensato y fructífero para comprender nuevos fenómenos, y todavía no había ningún fenómeno que, en su opinión, refutara claramente la teoría del flogisto. Y hay historias similares que contar sobre cada uno de los otros científicos brillantes y dedicados que se negaron a abandonar la teoría del flogisto, algunos de los cuales conoceremos en breve.
4.2 Agua
Un momento decisivo en la competencia entre la teoría del oxígeno y la teoría del flogisto fue el argumento de Lavoisier de que el agua no era un elemento en absoluto, sino un compuesto de oxígeno e hidrógeno. Vale la pena señalar la ironía de la situación. Nadie sabía mejor que Priestley cómo hacer oxígeno (aire desflogistizado), y el hidrógeno (llamado aire inflamable) había sido descubierto y estudiado en 1766 por su compatriota y colega flogistonista Henry Cavendish (1731-1810), dejando caer piezas de metal en ácidos[9]. Cavendish también descubrió cómo hacer explotar esos dos aires para hacer agua, un experimento que Priestley repitió con éxito. Fueron Priestley y Cavendish (a través de su amigo Charles Blagden) quienes le enseñaron a Lavoisier cómo hacer estas cosas. Aún así, dice la historia común, fue Lavoisier a quien se le ocurrió la interpretación correcta de qué eran estos gases y qué sucedió cuando reaccionaron entre sí. De hecho, no hay mejor caso que la composición del agua para ilustrar la sorprendente fuerza de la teoría del flogisto. Cavendish y Priestley pensaron durante un tiempo que el hidrógeno, o "aire inflammable" como lo llamaban, era puro flogisto, expulsado del metal por la acción del ácido (el metal se convertía así en cal y se disolvía en el ácido para formar una sal, Sal aquí designa una clase completa de sustancias químicas, incluida la sal común y muchas otras). Si se ponía una cal en el ácido, se disolvía sin producir aire inflamable, porque la cal no contenía flogisto.
Una versión más considerada de este punto de vista sirvió para explicar la formación de agua. El punto de vista posterior de Cavendish-Priestley fue que el aire inflamable era "agua flogística", es decir, agua que contenía un exceso de flogisto. En cuanto al oxígeno, o aire desflogistizado, eso era "agua desflogisticada". Cuando el agua flogisticada y el agua desflogisticada se combinan entre sí, el exceso y el déficit de flogisto cancelado y se produjo agua corriente. Para resumir: hubo (al menos) dos puntos de vista en competencia sobre la formación de agua, los cuales eran convincentes y autoconsistente:
(1) Hidrógeno + Oxígeno → Agua
(2) Agua flogisticada + Agua desflogística → Agua
El segundo relato aquí no es un cuento de hadas que los teóricos del flogisto simplemente fabricaron para evitar la refutación de su teoría por los hechos sobre la composición del agua. Cavendish y Priestley tenían buenas razones para pensar que el agua era un componente esencial de los gases. En el nivel más básico, esta idea se sugiere por el hecho de que los vapores se producen a partir de líquidos. ¿Podría la situación no ser similar también con otros tipos de gases? Lavoisier también estuvo de acuerdo en que los gases se producían a partir del agua y otros líquidos, mediante la adición de calórico (la materia del calor). Priestley (1788) se sentía completamente cómodo con "la suposición de que el agua entra en la constitución de todos los tipos de aire y es, por así decirlo, su base adecuada, sin la cual ninguna sustancia aeriforme puede subsistir[10]". Señaló que no conocía ninguna forma de producir aire inflamable sin agua, y supuso que “lo mismo puede ser cierto para cualquier otro tipo de aire, ya que el agua se usa en la producción de todos ellos[11]." Dentro de este marco de pensamiento, tenía mucho sentido pensar que la flogisticación o desflogisticación afectaría el proceso de aireación del agua, dando como resultado diferentes tipos de aires. Los filósofos históricamente bien informados se han esforzado por decir exactamente qué estaba mal con la postura de Priestley. Debemos resistir el impulso de decir "sabemos que estaba equivocado, porque el flogisto simplemente no existe", ya que eso solo plantea la pregunta de cómo sabemos eso. La queja de que no fue posible aislar el flogisto en su forma pura no tiene fuerza. Si siempre requiriéramos tal aislabilidad material, la ciencia se vería muy diferente, ya que tendríamos que renunciar a toda una gama de conceptos, desde los quarks hasta la energía. Y en el núcleo de la propia teoría de Lavoisier estaba el calor, la materia del calor, que tampoco era aislable en su forma pura. Tampoco está de más decir que el flogisto fue un concepto científico ilegítimo porque era inobservable.
Independientemente de cómo se defina “observable”, la ciencia hasta el día de hoy está llena de entidades inobservables que se postulan debido a necesidades teóricas (me vienen a la mente la materia oscura y las supercuerdas). Y no está claro que el flogisto no fuera observable; para los flogistonistas, el flogisto no solo era observable (en la llama que sale de la combustión, por ejemplo), sino incluso directamente manipulable (cuando se transfiere de una sustancia a otra, como en la fundición o en la producción de aire inflamable por la solución de metales en ácidos). Este sentimiento era evidente dentro de la Sociedad, esa notable asociación de personas científicas alrededor de Birmingham, que incluía a Priestley. En 1782, cuando la Revolución Química comenzaba a avanzar hacia su última fase, Matthew Boulton, socio comercial de James Watt, escribió al alfarero Josiah Wedgwood, maravillándose del nuevo experimento de Priestley en el que un calx se convertía en metal al "absorber" aire inflable (hidrógeno), que Priestley entonces consideró puro flogisto: “Hemos hablado durante mucho tiempo del flogisto sin saber de qué hablábamos, pero ahora el Dr. Priestley ha sacado a la luz el asunto. Podemos verter ese Elemento de un Vessell a otro, podemos decir cuánto de él es necesario medirlo con precisión para reducir un Calx a un Metal[12]... ".
Una queja relacionada contra el flogisto se refiere al peso. La versión cruda de la queja es que el flogisto es una sustancia “imponderable” (es decir, una sustancia ingrávida, no impensable) y, por lo tanto, no debería ser aceptada por la ciencia. Pero, ¿no es la física actual bastante optimista sobre las partículas ingrávidas, como los fotones? Y en la época del flogisto se postulaban impunemente otros imponderables, como el o los fluidos eléctricos, y no menos importante el calórico de Lavoisier. Otra versión de la proposición se centra en el aumento de peso en la calcinación: un metal gana peso al convertirse en calx, lo que no ocurriría si estuviera perdiendo algo, es decir, el flogisto. El aumento de peso se explica muy bien por la teoría del oxígeno, en la que la cal es metal combinado con oxígeno, por lo que obviamente es más pesado que el metal en sí mismo. Sin embargo, esto no funciona como una refutación de la teoría del flogisto, ya que hay formas de explicar el aumento de peso. No era necesario recurrir a la tan ridiculizada idea de que el flogisto tenía "ligereza[13]". Una explicación mucho más seria, presentada por Priestley y también por Richard Kirwan (1733-1812), fue que en la calcinación el metal se combinaba con agua, mientras perdía el flogisto. Cuando la cal se redujo de nuevo a metal, sedio el agua y absorbió el flogisto. Si no había una fuente externa de flogisto, el calx a veces lo tomaba del agua que estaba emitiendo, lo que significa que lo que emitía era agua desflogisticada (oxígeno, en términos de Lavoisier).
4.3 El problema con Lavoisier
Entonces podemos comenzar a ver cómo fue que Priestley pudo aferrarse a su teoría del flogisto, de una manera bastante racional. Lo que en realidad es más difícil de ver es por qué casi todos los demás deberían haberse inscrito en el puesto de Lavoisier o mantenerse en él durante un período de tiempo prolongado. Para liberar nuestro pensamiento de los viejos clichés sobre la Revolución Química, debo comenzar señalando cuán equivocado estaba Lavoisier, si lo juzgamos desde el punto de vista de la química y la física modernas. Como dice John McEvoy (1997, 22-23), es “un hecho simple” que ya “a fines del siglo XVIII, casi todas las afirmaciones teóricas importantes que Lavoisier hizo sobre la naturaleza y función del oxígeno eran insuficientes. " De manera similar, Robert Siegfried (1988, 35) afirma que “los supuestos centrales que habían guiado su trabajo de manera tan fructífera se demostraron empíricamente falsos alrededor de 1815”. Echemos un vistazo más de cerca. Tres pilares principales del sistema de química de Lavoisier marcaron claras desviaciones de la química anterior: la teoría de los ácidos, la teoría de la combustión y la teoría calórica. Los tres están claramente equivocados, desde el punto de vista de la química moderna, o incluso desde el punto de vista de la química del siglo XIX[14].
Incluso los entusiastas de Lavoisier más robustos admitirán fácilmente que su teoría de los ácidos estaba equivocada. Lavoisier dijo que todos los ácidos contenían oxígeno, pero los lavoisierianos sabían tan bien como cualquiera que no había evidencia de oxígeno en ciertos ácidos, incluido el ácido muriático (en términos modernos, ácido clorhídrico, HCl) y el ácido prúsico (ácido cianhídrico, HCN). Como dijo el químico de Oxford del siglo XX Harold Hartley (1971), "la rígida aceptación de esta doctrina" fue "responsable de tantas fantasías en las mentes de los químicos", incluida su incapacidad para reconocer el cloro como un elemento para 20 años después de su aislamiento por Scheele[15]. ¿Era esta teoría de los ácidos solo un desafortunado complemento no esencial del resto del sistema de Lavoisier, que podría descartarse con seguridad? Al menos el propio Lavoisier no lo creía así, como podemos vislumbrar por la forma en que nombró a su amado "oxígeno", que significa "generador de ácido[16]".
Aún más central para el sistema "antiflogístico" de Lavoisier era su teoría de la combustión. Sin duda, esta parte indiscutiblemente esencial del sistema de Lavoisier era correcta y todavía se conserva en la química moderna. Conceder eso sería participar en una amnesia que la historiografía prolavoisieriana ha orquestado cuidadosamente. De hecho, es bastante increíble que cualquier persona moderna pueda pensar: “la combustión es una combinación con oxígeno, eso es lo que causa la emisión de calor y luz, y Lavoisier descubrió todo eso". ¿Qué diablos tiene que ver el oxígeno con el calor y la luz? La explicación de Lavoisier de la producción de calor en la combustión era que era la liberación del fluido calórico del oxígeno gaseoso, el calórico responsable del estado gaseoso del oxígeno en primer lugar. Pero fue ampliamente reconocido por los contemporáneos de Lavoisier (e incluso por el mismo Lavoisier) que había serias dificultades con esta historia. Thomas Thomson (1773 1852), el principal químico escocés del período inmediatamente posterior a la Revolución Química, dio un resumen sereno y devastador de conocidas dificultades en su System of Chemistry, publicado por primera vez en 1802[17]. Estas dificultades incluyeron casos de combustión sin oxígeno en estado gaseoso (como la explosión de pólvora) y combustión sin oxígeno en absoluto. Thomson juzgó que "en general, no se puede negar que la teoría de Lavoisier no ofrece una explicación suficiente de la combustión". Thomson no abogaba por un regreso al flogisto, pero quería que la química avanzara más allá de Lavoisier, menos de 15 años después de la culminación de la Revolución Química.
También había un creciente descontento con la teoría calórica del calor de Lavoisier en términos más generales, particularmente en Londres, donde alrededor de 1800 había una notable concentración de defensores de la noción de que el calor era una forma de movimiento, incluidos el Conde Rumford, Humphry Davy, Thomas Young y Henry Cavendish. Es importante recordar que el calórico en el sistema de Lavoisier no era simplemente un dispositivo para explicar la liberación de calor en la combustión; más bien, era un elemento esencial en su cosmología, por ejemplo para explicar los tres estados de la materia. Lavoisier consideró claramente al calórico como una piedra angular de su sistema químico, colocándolo (junto con la luz) en la parte superior de su lista de elementos químicos y dedicando todo el primer capítulo de su libro de texto definitivo de nueva química para elucidar la naturaleza y el papel del calórico. Era un sistema hermoso y sensato, pero solo en la medida en que lo era la teoría del flogisto. Ambas teorías son igualmente erróneas, desde el punto de vista moderno.
Reconociendo que el debate no era una simple cuestión de verdad y falsedad, varios filósofos e historiadores de la ciencia han intentado afrontar el desafío de explicar por qué la gran mayoría de los químicos se pasaron al lado de Lavoisier. El artículo clásico de Alan Musgrave sobre el tema analiza y rechaza varios intentos comunes (1976[18]). No es el caso de que la nueva química de Lavoisier se estableció por inducción a partir de observaciones. Es igualmente incorrecto decir que la teoría del flogisto fue simplemente refutada por hechos como el aumento de peso de los metales cuando se oxidan (o se calcinan). Lo que puede ser más atractivo pero igualmente falaz es la noción de que la teoría de Lavoisier ganó porque era inherentemente más simple que la teoría del flogisto. La versión más cruda de esta idea, que Musgrave denomina “simplicismo” (o convencionalismo) en su crítica, dice que la teoría del flogisto complica innecesariamente las cosas al postular una sustancia inobservable, el flogisto; este argumento ignora el hecho de que Lavoisier tuvo que postular una sustancia igualmente inobservable, la calórica.
Después de descartar estas explicaciones, Musgrave propone que el factor crucial fue que el programa de investigación del oxígeno fue más progresivo, con "progreso" como se define en la filosofía de Imre Lakatos[19]. Musgrave sostiene que era racional para los químicos abandonar la teoría del flogisto porque después de cierto punto dejó de hacer nuevas predicciones exitosas; solo continuó formulando hipótesis ad hoc, excusas inventadas para proteger una teoría fallida[20]. Desafortunadamente, no creemos que esta explicación lakatosiana funcione, como explicaremos con más detalle. Musgrave afirma: "Entre 1770 y 1785, el programa de oxígeno demostró claramente su superioridad al flogistonismo: se desarrolló de manera coherente y cada nueva versión fue teórica y empíricamente progresiva, mientras que después de 1770 el programa del flogisto no hizo ninguna de las dos cosas". Encuentro esta afirmación difícil de fundamentar, por atractiva que sea. La idea de que el progreso del programa de flogisto terminó en 1770 (mientras que el programa de oxígeno continuó en su suave progreso) se contradice con las propias declaraciones de Musgrave anteriormente en el artículo: “Mientras Lavoisier fallaba, Priestley estaba teniendo un gran éxito con la versión de 1766 del flogistonismo… el experimento más impresionante de todos se produjo a principios de 1783 ". Esta fue la confirmación de la predicción flogistonista de que los cálices se reducirían a metales por calentamiento en aire inflamable. Para sostener la tesis de Musgrave de manera convincente, necesitamos encontrar predicciones novedosas y exitosas que el programa de investigación de Lavoisier hizo después de que el programa del flogisto dejó de producirlas, y eso es 1783, no 1770. ¿Dónde están estas predicciones? ¿Estamos pensando en la predicción de que la oxidación del aire inflamable (hidrógeno) produciría un ácido? ¿O la predicción de que el ácido muriático (clorhídrico) se descompondría en oxígeno y el “radical muriático”?
Tras el fracaso de todos estos intentos por defender la racionalidad del consenso a favor de Lavoisier, la historia se complica más. A menudo se persiguen dos líneas de pensamiento comunes: defender la presunta racionalidad de la Revolución Química apelando a una versión complicada del simplicismo, o desviar la atención de la preocupación filosófica tradicional por la racionalidad buscando explicaciones sociales. Consideraremos estas líneas de pensamiento y por qué no las seguimos, ya estará claro para entonces a partir de la declaración de nuestra propia posición. Por ahora, sin embargo, permítanos continuar con la implicación de la conclusión a la que vamos a llegar después de la deliberación completa: la teoría del flogisto fue destruida prematuramente.
4.4 ¿Podría ser el agua un elemento?
La carga del juicio es una responsabilidad de actuar: si realmente creemos que la teoría del flogisto fue descartada prematuramente, debemos considerar qué se podría haber logrado manteniéndola. Nuestro objetivo principal no es la historia contrafactual de "qué pasaría si". En última instancia, estamos abogando por un tipo de didáctica más activista, en la que realmente abramos la posibilidad de revivir la línea de pensamiento injustamente descartada y ver qué sale de ella. Lo que buscamos es una visión completa: queremos saber qué contribuciones al conocimiento científico hizo la teoría del flogisto, qué contribuciones podría haber hecho si se hubiera mantenido más tiempo y qué contribuciones podría hacer aún si se reviviera. Si todas esas categorías de contribuciones se han perdido o se han perdido debido al abandono prematuro de la teoría del flogisto, entonces deberíamos recuperarlas, imaginarlas y crearlas. Si objetara que tal empresa no es ni historia ni filosofía ni ciencia real, que así sea: la llamo “ciencia complementaria”. Nuestro objetivo es dar una función novedosa a la historia y la filosofía de la ciencia, sin negar sus funciones tradicionales. Y, por supuesto, no estamos proponiendo eliminar el oxígeno o la tradición de la química moderna descendiente de Lavoisier, incluso si tal cosa fuera posible. No, toda la empresa es pluralista, como explicaremos más adelante en términos más generales. Hay varias preguntas que debemos hacer. (1) ¿Hubo algún conocimiento que los científicos perdieron cuando rechazaron el sistema flogistonista? (De aquí en adelante diré “sistema” en lugar de “teoría”, para enfatizar que hay más que teoría involucrada) En otras palabras, ¿hubo algo bueno que hizo el sistema flogistonista que el sistema oxigenista no pudo hacer? ¿hacer? Kuhn pensó que, por lo general, existía tal pérdida de conocimiento cuando ocurre una revolución científica; esto se ha denominado "pérdida de Kuhn" en su honor. (2) ¿Hubo algún conocimiento que pudiera provenir de mantener el sistema flogistonista, cuyo desarrollo fue retrasado o impedido por su desaparición? (3) ¿Hubo un efecto beneficioso de tener presentes tanto los sistemas flogistonista como oxigenista, en términos de lo que se produjo por las interacciones entre ellos? (4) ¿Habría habido más interacciones beneficiosas entre el sistema oxigenista y el sistema flogistonista, si se hubiera mantenido este último?
Intentaremos responder estas preguntas, pero aquí hay algunos pensamientos preliminares para estimular su imaginación. Aunque muchas de las explicaciones de los fenómenos químicos dadas en el sistema flogistonista fueron incorporadas con éxito al sistema oxigenista, eso no siempre fue posible. Por ejemplo, la teoría del flogisto dio una buena explicación de por qué todos los metales tenían propiedades similares, y muy poco sobre eso podría decirse en la teoría de Lavoisier. La identificación del flogisto con la electricidad (negativa) fue una prometedora vía de pensamiento cerrada por la desaparición del flogisto. Si bien los teóricos del flogisto a menudo son criticados por no haber prestado suficiente atención al equilibrio de pesos en las ecuaciones químicas, el flogisto sirvió como un recordatorio útil de que no todo en química puede explicarse por pesos. El eminente químico inglés William Odling opinó en 1871 que el flogisto había sido un claro precursor de la energía potencial química. Estas ideas deberían al menos darnos una pausa y mucho para pensar[21].
Nuestra visión idiosincrásica de la revolución química hará que mucha gente se sienta incómoda. Si el rechazo de la teoría del flogisto fue prematuro e injustificado, también lo fue el rechazo de la idea de que el agua es un elemento. ¿Pero seguramente el agua no es un elemento? Si crees en algo tan extravagante, todo el peso de la ciencia moderna te aplastará. Pero, ¿no podría haber realmente un sistema científico sensato en el que el agua sea un elemento? La respuesta fácil es: "Sí, y se le llamó el sistema flogistónico", o, en realidad, todos los sistemas científicos antes de la Revolución Química. La pregunta más difícil, sin embargo, es si existe un sistema de ciencia basado en el agua elemental en el que nosotros, aquí y ahora, podamos creer plausiblemente, o al menos trabajar con y sacar provecho. Al considerar esta cuestión, debemos, nuevamente, resistir la tentación de decir “por supuesto que ahora sabemos que el agua es un compuesto de hidrógeno y oxígeno; cualquier teoría que diga lo contrario es simplemente falsa y no vale la pena considerarla ”.
Solo pensamos así porque somos prisioneros de una cosmovisión científica que se basa en premisas como la naturaleza compuesta del agua. El desafío verdaderamente pluralista es preguntarnos si podríamos salirnos de esa cosmovisión y encontrar otra que no se base en la naturaleza compuesta del agua; y, si lo hubiera, si su desarrollo tendría algún beneficio. Como dice Léna Soler[22], la pregunta general es si los resultados científicos bien establecidos son inevitables o contingentes: ¿es “posible que haya una ciencia que lo sea? ... tan exitoso y progresista como el nuestro pero radicalmente diferente en contenido”?.
Un conocimiento terminológico, trivial en sí mismo, hará que esta perspectiva parezca infinitamente más plausible. Cuando hablamos de "elementos" en la ciencia moderna, por ejemplo, cuando decimos que el oxígeno es un elemento químico, no nos referimos en última instancia a cuerpos simples que no se pueden descomponer más, o incluso a cuerpos que aún no se han descompuesto. Tales ideas ingenuas de los elementos químicos tuvieron que ser rechazadas en la ciencia posterior, al igual que la opinión de que el agua era un elemento y no podía descomponerse más.
Según la ciencia moderna, los átomos y moléculas de oxígeno, hidrógeno o agua, todas y cada una de las sustancias químicas, están formadas por otras partículas más simples, como neutrones, electrones y protones. Si nos tomamos muy en serio la física moderna, ni siquiera un electrón es una partícula "simple"; no es una “partícula” en absoluto en su sentido ordinario, sino un paquete de energía que exhibe dualidad onda-partícula, o ni siquiera eso, sino algún estado fluctuante del campo cuántico. No importa eso, el punto es que "El agua es un elemento" es realmente tan erróneo como "El oxígeno es un elemento". De modo que no hay razón para sentirse tan desesperadamente arrepentido al considerar los méritos potenciales de una teoría que incluye agua elemento, como tampoco al considerar los méritos de una teoría que incluye oxígeno elemental. La verdadera pregunta es qué significa "elemento" en un sistema de química, en términos de sus funciones. Cuando Priestley y Cavendish sostuvieron que el agua era un elemento, claramente lo entendieron como una sustancia que podía modificarse, mediante la adición o sustracción de flogisto, por ejemplo. Ciertamente podemos considerar si hay ideas potenciales que podamos obtener de esa forma de pensar.
Tomemos el caso del agua en la Revolución Química como un recordatorio de la pobreza de la visión única. La historia del flogisto ilustra cómo se puede perder el conocimiento real y potencial si insistimos en tener una sola forma de perseguirlo.
4.5 Por qué debería haber vivido Phlogiston
Habiendo planteado algunas dudas y preguntas sobre el resultado de la Revolución Química, ahora daremos una exposición completa y libre de nuestros puntos de vista. Aquí asumimos una buena cantidad de conocimientos previos y discutimos asuntos con poca atención a los límites disciplinarios o preocupaciones sobre objeciones que puedan provenir de varios especialistas; luego, trataremos las objeciones anticipadas y de posicionar puntos de vista con respecto a la literatura existente. Una valoración sistemática de la situación probatoria en la elección entre oxígeno y flogisto. El veredicto de esa tasación será en la línea ya indicada: no existían pruebas concluyentes para rechazar la teoría del flogisto en favor de la teoría de Lavoisier. Esto plantea una cuestión de explicación histórica: ¿por qué, entonces, los químicos hicieron esa elección injustificada?, los químicos de la época no llegaron a un acuerdo simple, rápido y universal a favor de Lavoisier. Aún así, admitiendo que una clara mayoría finalmente rechazó el flogisto, intentar explicar por qué se llegó a esa decisión colectiva, haciendo referencia a una tendencia más amplia y de más largo plazo subyacente a la Revolución Química, a saber, el advenimiento del "composicionismo". Dar una explicación no significa defender lo que se ha explicado. Después de todas las evaluaciones y explicaciones, analizar mejor el destino del flogisto y se preguntará qué beneficios se podrían haber derivado de retenerlo en química.
4.6 Phlogiston contra oxígeno
¿Había suficiente justificación científica para el rechazo de la química basada en el flogisto a favor de la química basada en el oxígeno de Lavoisier? ¿Hubo suficiente evidencia para apoyar esa decisión? Nuestra sensación es que todavía no ha habido una evaluación verdaderamente sistemática de este asunto. Ahora, esa es una afirmación grandiosa sobre un tema que se ha estudiado tan a fondo, así que necesitamos explicar lo que tenemos en mente con más cuidado. En primer lugar, gran parte de la literatura histórica existente sobre la revolución química no se ocupa de las cuestiones de la justificación. Y la mayoría de los análisis filosóficos se han llevado a cabo desde perspectivas particulares, cada una de las cuales descuida u oscurece ciertos aspectos de la situación, como lo explicaremos más adelante. Por tanto, empezaremos por esbozar un marco de análisis más cómodo.
4.7 Evaluación de sistemas de práctica
Al menos en las tradiciones anglófonas, los análisis filosóficos de la ciencia se han visto excesivamente limitados por el hábito común de ver la ciencia como una colección de proposiciones, centrándose en el valor de verdad de esas proposiciones y las relaciones lógicas entre ellas. El principal tema de discusión en filosofía de la ciencia han sido las teorías como cuerpos organizados de proposiciones. Esto ha llevado al descuido de la experimentación y otras dimensiones no verbales y no proposicionales de la ciencia en los análisis filosóficos. Muchos historiadores, sociólogos y filósofos han señalado este problema, pero hasta ahora no se ha acordado un marco filosófico alternativo claro que proporcione un lenguaje para análisis más completos de la práctica científica. Un estudio serio de la práctica científica debe preocuparse por lo que realmente hacemos en el trabajo científico. Esto requiere un cambio de enfoque de propuestas a acciones, iniciando con el reconocimiento de que todo el trabajo científico, incluida la teorización pura, consiste en acciones —físicas, mentales y operaciones de "papel y lápiz", para ponerlo en términos de Percy Bridgman[23]. Por supuesto, todas las descripciones verbales que hacemos del trabajo científico deben ponerse en proposiciones, pero debemos evitar el error de prestar atención únicamente a los aspectos proposicionales de las acciones científicas.
Proponemos enmarcar nuestros análisis en términos de “sistemas de práctica (científica)” que se componen de “actividades epistémicas[24]”. (También usaré la frase "sistema de conocimiento" de manera intercambiable con "sistema de práctica", especialmente cuando parece que no hay peligro de olvidar que el conocimiento está arraigado en la práctica). Una actividad epistémica es una actividad más o menos conjunto coherente de operaciones mentales o físicas que están destinadas a contribuir a la producción o mejora del conocimiento de una manera particular, de acuerdo con algunas reglas discernibles (aunque las reglas pueden no estar articuladas). Una parte importante de mi propuesta es tener en cuenta los objetivos que los científicos intentan alcanzar en cada situación. La presencia de un objetivo identificable (incluso si no está expresamente articulado por los propios actores) es lo que distingue las actividades de los meros sucesos físicos que involucran cuerpos humanos, y la coherencia de una actividad se define porque también la actividad logra alcanzar su objetivo. Los tipos comunes de actividades epistémicas incluyen medición, predicción y prueba de hipótesis. Algunas actividades epistémicas son principalmente mentales. Existe la práctica teórica, y en química consiste en actividades como la clasificación, el equilibrio de ecuaciones y el modelado de estructuras moleculares. En realidad, la mayoría de las actividades epistémicas son tanto mentales como físicas a la vez. Cuando empezamos a pensar en el trabajo científico como una colección de actividades, algo inmediatamente obvio es la gran variedad de tipos de actividades epistémicas en las que participan los científicos[25]. Aquí hay una lista parcial de tipos de actividades epistémicas: describir, predecir, explicar, formular hipótesis. , probar, observar, detectar, medir, clasificar, representar, modelar, simular, sintetizar, analizar, abstraer, idealizar. Las actividades epistémicas normalmente no ocurren, y no deberían, ocurrir de forma aislada. Más bien, cada uno tiende a practicarse en relación con otros, constituyendo todo un sistema.
Un sistema de práctica está formado por un conjunto coherente de actividades epistémicas realizadas con miras a lograr ciertos objetivos. Por ejemplo, Lavoisier creó un sistema de química cuyas actividades incluían recolectar gases producidos por reacciones químicas, medir los pesos de los ingredientes y productos de las reacciones, quemar sustancias orgánicas con fines analíticos y clasificar compuestos según sus composiciones[26]. Los objetivos generales de este sistema incluían determinar la composición de varias sustancias y explicar las reacciones químicas en términos de la composición de las sustancias. De manera similar, como ocurre con la coherencia de cada actividad, son los objetivos generales de un sistema de práctica los que definen lo que significa que el sistema sea coherente. La coherencia de un sistema va más allá de la mera coherencia entre las proposiciones involucradas en sus actividades; más bien, la coherencia consiste en varias actividades que se conjugan de manera eficaz hacia la consecución de los objetivos del sistema.
La coherencia se presenta en grados y formas diferentes, y es necesariamente un concepto menos preciso que la coherencia, que viene bien definida a través de axiomas lógicos. Puede parecer difícil hacer una clara distinción entre una actividad epistémica y un sistema de práctica, y esto es intencional. La distinción es solo relativa y depende del contexto. En cada situación en la que estudiamos un cuerpo de práctica científica, propongo llamar sistema al objeto general; cuando queremos estudiar más de cerca aspectos más específicos de ese sistema, podemos analizarlo en diferentes actividades subordinadas. Lo que tomamos como un sistema completo en una situación dada puede verse como una actividad constitutiva de un sistema más amplio, y lo que vemos como una actividad constitutiva en una situación dada puede, en una situación diferente, analizarse como un sistema completo compuesto por otras actividades. (Cuando introdujimos inicialmente la noción de actividad epistémica hace dos párrafos, utilicé el expediente de decir que una actividad se compone de operaciones; la terminología de "operación" es estrictamente innecesaria, aunque es conveniente cuando queremos mantener tres niveles de descripción a la vista todos a la vez).
De esta manera, mi marco es aplicable a todos los niveles y se puede acercar y alejar para adaptarse a cualquier nivel en el que queramos centrarnos. En cada punto de enfoque, llamamos a la práctica general "sistema" y sus constituyentes "actividades" (y sus constituyentes "operaciones"), sin la intención de pegar esas etiquetas categóricas a nada de forma permanente. No me propongo profundizar en la metafísica de la acción, pero será útil subrayar la naturaleza no reductiva de la relación entre los niveles de descripción aquí. La estructura de acciones y procesos no es atomista de manera reductiva, a diferencia de la estructura de cosas y enunciados[27]. Cada actividad epistémica se puede analizar en sí misma como un sistema de actividades, pero las actividades "componentes" no son necesariamente más simples que la actividad "total" en un sentido absoluto, y el análisis puede continuar indefinidamente. Por ejemplo, tome el análisis de combustión de una sustancia química.
Esto puede analizarse como compuesto por varias otras actividades: quemar la sustancia objetivo; absorber los productos de combustión utilizando otros productos químicos; pesar con una balanza; hacer cálculos porcentuales; etc. Y esas actividades componentes en sí mismas consisten en otras actividades; por ejemplo, la actividad de pesar con balanza consiste en colocar muestras y pesas en platillos de balanza, leer el número de la balanza, etc. Ahora puede parecer que estamos llegando a actividades cada vez más simples a medida que continuamos en nuestro análisis de acciones, con suerte para llegar al fondo de las operaciones atómicas. Pero nos estamos olvidando de algo menos conveniente. La actividad de pesar con balanza también incluye una actividad de certificación, para respaldar nuestro supuesto de que lo que estamos manejando son los pesos estándar correctos; sin eso, toda la actividad de pesar con una balanza se vuelve incoherente. Esta actividad de certificación puede consistir en pedir las pesas a un proveedor confiable, o compararlas con un conjunto de pesas más confiable, o compararlas con ciertos fenómenos naturales (por ejemplo, el peso de un cierto volumen de agua a una cierta temperatura).
Sea cual sea la opción que escojamos, está claro que esta actividad componente no es más sencilla en ningún sentido claro que la actividad principal de pesar con balanza. La relación entre varias actividades epistémicas es, en última instancia, no reductiva y reticular, aunque en muchas situaciones podemos obtener conocimientos útiles al analizar una actividad en sus componentes aparentes. No hay un nivel mínimo de descripción ni un final claro para el proceso de análisis de actividad. Más bien, el análisis debe realizarse donde sea y en la medida en que sea productivo.
Antes de comenzar a aplicar los conceptos de actividad epistémica y sistema de práctica al caso que nos ocupa, algunas observaciones más generales ayudarán a aclarar aún más el marco. Primero, el enfoque en las actividades y sus objetivos arroja nueva luz sobre la antigua preocupación filosófica por las proposiciones y su verdad. Por supuesto, las proposiciones son importantes en la ciencia, incluidos los enunciados de observación, las leyes empíricas y los principios teóricos. Sin embargo, lo realmente importante es comprender cómo funcionan dentro de varias actividades epistémicas. La evaluación de la exactitud de las declaraciones es sin duda una de las actividades epistémicas más importantes; sin embargo, los estándares de corrección pueden variar fácilmente de un sistema a otro. Si estamos hablando de una "Verdad con una T mayúscula por su palabra en inglés“ que no depende en absoluto del sistema de práctica en el que uno trabaja, es dudoso que haya actividades científicas reales que se ocupen de ella. El éxito de cada actividad o sistema debe juzgarse en primer lugar en términos de qué tan bien logra los objetivos que se fija; además, podemos emitir juicios sobre el valor de los objetivos mismos. Pero habrá muy pocas ocasiones en las que la "Verdad" sea un objetivo operable o un estándar de juicio en la ciencia[28].
Finalmente, resultará instructivo establecer una comparación y un contraste entre los sistemas de práctica y algunas otras nociones ya corrientes en la literatura de la historia y la filosofía de la ciencia. La comparación más obvia es el concepto kuhniano de paradigma. A pesar de algunas claras similitudes, creo que también hay algunas diferencias significativas, suficientes para justificar la articulación de un nuevo concepto. Como admitió libremente Kuhn, utilizó el término "paradigma" en dos sentidos principales[29]. El primero, de “ejemplar”, no se corresponde con mi sentido de “sistema”. El segundo sentido, la “matriz disciplinaria”, es similar a mi concepto, pero por dos razones principales no lo encuentro útil. Primero, creo que necesitamos un concepto que sea más definido y ordenado que la matriz disciplinaria kuhniana, que incorpore todo tipo de elementos que van desde los principios metafísicos fundamentales hasta las estructuras institucionales, sin una indicación definitiva de cómo todo se mantiene unido. La caracterización de Hacking (1992) de la práctica de laboratorio también tiene una dificultad similar, especificando 15 tipos diferentes de elementos que entran en la práctica experimental sin elaborar cómo se combinan e interactúan entre sí[30]. Además, el concepto de paradigma de Kuhn está demasiado ligado a su insistencia en que un paradigma tiene y debe disfrutar del monopolio de toda una disciplina científica en las fases normales de la ciencia; tenemos objeciones tanto descriptivas como normativas a esa presunción de monopolio. Otra comparación interesante es con las "formas de conocimiento" de John Pickstone, que creo que son tipos de sistemas de práctica (o tipos de sistemas[31]). Un tipo de sistema es una clase que consta de varios sistemas, todos los cuales comparten algunas actividades básicas.
Un tipo de sistema no es un sistema real, ni siquiera uno muy general, sino una caracterización incompleta de un sistema con espacios en blanco que se dejan conscientemente por completar (y de manera similar para el tipo de actividad, una noción que utilicé brevemente anteriormente). Dependiendo de cómo los espacios en blanco están llenos, tenemos diferentes instancias del tipo. Un tipo de sistema define una clase de sistemas que consta de estas instancias[32].
4.8 Campos de problemas
Habiendo aclarado nuestro marco de análisis, permítanos pasar a la tarea que nos ocupa: comparar los méritos relativos de los sistemas flogistonista y oxigenista de la química tal como existían a fines del siglo XVIII. Al intentar responder a estas preguntas, es importante mantener la cabeza clara sobre el juicio de quién estamos considerando. Hay varios puntos de vista a considerar: qué pensó cada uno de los lados en competencia en la disputa y qué pensamos nosotros mismos como investigadores académicos. En cuál de estos juicios nos enfocamos, depende de nuestro propósito al hacer la investigación. Nuestro propósito actual es evaluar si hubo buenas razones para abandonar el sistema flogistonista, no solo si los científicos del pasado lo pensaron así. Por lo tanto, debemos dar nuestros propios juicios, aunque sería un error ignorar por completo los juicios de los actores históricos.
Trataremos de ser comprensivos en nuestra valoración, porque lo que hemos visto hasta ahora indica que nunca hubo un simple “factor decisivo” aislado o una falla fatal en ninguno de los lados que resolvió, o debería haber arreglado, la discusión. Eso significa que probablemente hubo una serie de factores importantes relevantes para la decisión, todos los cuales deben considerarse cuidadosamente y sopesarse entre sí.
Entonces, comencemos por preguntarnos, de manera muy general: ¿cuáles fueron los problemas científicos importantes que enfrentaron los sistemas flogistónico y oxigenista, y qué tan bien los resolvió cada sistema? Responder a estas preguntas requerirá que abordemos otras dos preguntas: según qué estándares, o valores epistémicos, deberían evaluarse las soluciones a esos problemas; y ¿cómo exactamente debería aplicarse cada valor epistémico en las situaciones concretas que se están considerando?
Todas estas preguntas fueron planteadas muy claramente por Kuhn (1970, 1977) en sus discusiones sobre la elección de paradigmas frente a la inconmensurabilidad. En La estructura de las revoluciones científicas, uno de los puntos principales de Kuhn con respecto a la inconmensurabilidad fue que los diferentes paradigmas tienen diferentes listas de problemas que consideran legítimos e importantes. Este elemento más indiscutible de inconmensurabilidad a menudo se dejaba de lado en las emocionantes discusiones sobre sus otros aspectos, y parece que el propio Kuhn abandonó la preocupación por los campos problemáticos en las discusiones basadas en referencias de inconmensurabilidad que dio más adelante en su vida. Pero no vemos cómo podemos siquiera iniciar un debate razonado sobre la elección de la teoría o la elección del paradigma sin identificar los campos de problemas reconocidos por la química, que era “explicar las cualidades de las sustancias químicas y los cambios que estas cualidades experimentaron” durante las reacciones químicas. Más específicamente, los flogistonistas buscaron explicar las propiedades clave de muchas sustancias en términos de los "principios", particularmente el flogisto, que entraron en su composición. Un caso destacado fue la explicación de por qué los metales (que eran compuestos para los flogistonistas) tenían un conjunto de propiedades comunes. En realidad, con el inicio de la Revolución Química, esto ya no era un problema de investigación en el paradigma del flogisto, ya que se aceptaba casi como sentido común que los metales tenían sus propiedades metálicas comunes (incluyendo brillo, maleabilidad, ductilidad, conductividad eléctrica) debido al flogisto. El lado oxigenista parece haber rechazado no tanto esta respuesta como la pregunta misma; la química recuperó este tramo de territorio solo en el siglo XX. También hubo algunos otros problemas desatendidos por los oxigenistas que fueron considerados importantes por algunos flogistonistas, aunque no todos los flogistonistas estaban igualmente preocupados por ellos. Estos incluyeron varios problemas en mineralogía, geología, meteorología y la circulación de nutrientes en el medio ambiente.
Por otro lado, había algunos problemas considerados mucho más importantes por los oxigenistas que por los flogistonistas. Todos los químicos notaron los fenómenos térmicos y varios flogistonistas intentaron dar alguna explicación sobre la naturaleza del calor, pero fue Lavoisier, basándose en el trabajo de Joseph Black sobre el calor latente, quien realmente llevó el calor (calórico) de manera centralizada a la química. Estrechamente relacionadas con la teoría del calor estaban las cuestiones relativas a lo que ahora llamamos cambios de estado, sobre los que Lavoisier tenía una teoría muy definida, a la que concedió un lugar muy destacado en su sistema de química.
Un caso similar es la química de las sales. Esta era una preocupación común de la química del siglo XVIII, pero la teoría del flogisto tenía relativamente poco que ofrecer aquí. En contraste, esta área de investigación era muy prometedora para la nueva química sobre la base de la noción de Lavoisier de que "el oxígeno formaba el pegamento o enlace de la unión dualista entre el ácido y la base para formar sales". Lavoisier mostró su entusiasmo por esta línea de investigación al dedicarle un tercio de sus Elementos de química.
Habiendo examinado el campo de problemas de la Revolución Química, ahora podemos abordar nuestra pregunta principal: ¿cómo se comparan los rendimientos de los dos lados? El primer juicio es que todos los problemas enumerados en la tabla 1.1 eran importantes y valiosos. Ahora, cuando hacemos tales juicios, pasamos por alto los juicios de los actores históricos sobre lo que era importante. Los historiadores tenderán a desconfiar de tales juicios, pero no es uno que podamos evitar. Los filósofos que deseen hacer una evaluación epistémica ciertamente no pueden evitarlo, e incluso los historiadores deben juzgar sobre qué vale la pena escribir. Simplemente seguir los juicios de los actores históricos no es una solución, especialmente en casos como este, en los que científicos importantes en ese momento no estaban de acuerdo entre sí. La opción más perniciosa es seguir simplemente el juicio del ganador histórico.

Aceptando que todos los problemas considerados importantes por cualquiera de las partes eran dignos (al menos a prima vista), podemos preguntarnos qué tan bien lo hizo cada una de las partes para resolverlos. De manera bastante predecible, y tal como dijo Kuhn, cada lado tendía a brindar buenas soluciones a los problemas que consideraba importantes, y no tan buenas soluciones a otros problemas; eso no nos da mucha base para preferir un lado al otro. Nos parece que hubo un mayor número de problemas importantes tratados por el sistema flogistonista pero no por el sistema oxigenista. En igualdad de condiciones, esa circunstancia tendería a recomendar el sistema flogistonista sobre el sistema oxigenista. Posiblemente, lo más importante es si alguna de las partes lo hizo claramente mejor en la solución de los problemas comunes, que ambas partes reconocieron como importantes. En la Revolución Química no faltaron los problemas comunes, pero cada lado pensó que lo estaba haciendo bastante bien en todos ellos, y bastante mejor que el otro.
Los defensores de los dos sistemas ofrecían soluciones sustancialmente diferentes a problemas comunes y se entendían bastante bien, pero no estaban de acuerdo con la evaluación de las cualidades relativas de esas soluciones. Esto nos lleva a la cuestión de los criterios de juicio, o los valores epistémicos que operaban en los debates.
Para explicar todos los fenómenos principales que produjeron y observaron, incluso si las explicaciones se volvieron engorrosas en los casos más difíciles. No es que ninguna de las partes haya fallado en reconocer la conveniencia de la simplicidad o la integridad, sino que hubo claras diferencias en el grado de énfasis o preocupación con esos valores en competencia. Un buen ejemplo para ilustrar este punto es la calcinación y reducción de metales. Lavoisier y sus colegas consideraron el calx rojo de mercurio, que Priestley había utilizado inicialmente para producir aire desflogistizado, como el caso paradigmático que muestra que la calcinación y la reducción eran procesos de oxidación y desoxidación. El mercurio podría convertirse en este cáliz rojo calentándolo con aire ordinario; la cal se podría convertir de nuevo en metal simplemente por un mayor grado de calor (producido por una lente ardiente grande), produciendo oxígeno y sin producir o absorber ninguna otra sustancia detectable. Esta maravillosa exhibición de oxidación y reducción fue citada una y otra vez por los lavoisierianos. Priestley protestó: "Pero este es el caso de la calx particular de este metal". En su opinión, los lavoisierianos estaban distorsionando todo el panorama al centrarse en un caso excepcional. Otros metales se comportaron de manera diferente; Priestley señaló que ningún calcio de hierro podría revivirse "a menos que se caliente en aire inflamable, que ingiere con entusiasmo, o en contacto con alguna otra sustancia que se supone que contiene flogisto". Incluso para el mercurio, había otro tipo de calx que no puede revivirse completamente con ningún grado de calor, pero puede revivirse en aire inflamable, que absorbe, o cuando se mezcla con carbón vegetal, revestimientos de hierro u otras sustancias se supone que contiene flogisto.
Esta divergencia entre simplicidad e integridad también jugó un papel importante en los debates sobre combustión. Las dificultades de la teoría de la combustión de Lavoisier, que discutimos brevemente, merecen una atención detallada aquí, especialmente porque tienden a descuidarse incluso en tratamientos históricos bien informados. Como antecedente, recordemos que Lavoisier entendió la combustión como una descomposición del oxígeno gaseoso en "oxígeno base" y calórico, combinándose el oxígeno base con la sustancia combustible y liberando el calórico. El calor sensible generado en la combustión provenía del oxígeno gaseoso, y era esencial que el oxígeno que permitía la combustión estuviera en estado gaseoso para empezar, ya que era la abundancia de calórico combinado lo que ponía una sustancia en estado gaseoso. La producción de luz en combustión se explicó de manera similar, aunque de manera más vaga.
Permítanme volver al resumen de las objeciones de Thomas Thomson a la teoría de la combustión de Lavoisier (Thomson 1802). Thomson estimó que, siguiendo el punto de vista de Lavoisier, "uno podría suponer naturalmente que cuando el producto [de la combustión] es un gas, todo el calor y la luz que existían en el oxígeno gaseoso serían necesarios para mantener el estado gaseoso del producto". Pero, por ejemplo, cuando se quema carbón vegetal, el producto es un gas, pero la combustión todavía produce una gran cantidad de calor y luz. Según Thomson, Lavoisier era consciente de este problema, pero no proporcionó una solución convincente. Thomson también notó un problema inverso: “Uno podría suponer naturalmente que en todos los casos de combustión el oxígeno empleado debe estar en el estado de un gas. Pero esto está muy lejos de ser el caso”. Por ejemplo, "una combustión muy rápida" tiene lugar cuando se vierte ácido nítrico sobre ciertos aceites, pero el oxígeno solo entra en esa reacción en estado líquido, no gaseoso. O considere la explosión de la pólvora, que ocurre sin la ayuda del gas de oxígeno ambiental, estando el oxígeno presente en estado sólido en el nitro (salitre) contenido en la pólvora misma.
Thomson también señaló que a partir de la teoría de Lavoisier uno esperaría naturalmente que se emitiera calor y luz durante la condensación de otros gases además del oxígeno: pero esto nunca sucede a menos que se trate de oxígeno. Por ejemplo, cuando los gases de hidrógeno y nitrógeno se combinan, no se emite luz ni calor; el gas amoniaco y el gas ácido clorhídrico se combinan para formar una "sal de hormigón", que produce muy poco calor y nada de luz. Thomson también señaló que hubo una emisión de una gran cantidad de calorías y luz (es decir, combustión, según todas las apariencias) en algunas reacciones que no involucraron oxígeno (ni gases) como reactivos: por ejemplo, cuando el azufre se combina con ciertos metales, y cuando el fósforo y la cal se combinan entre sí.
Tales problemas no movieron a Lavoisier y sus colegas a modificar sus doctrinas, por no mencionar a abandonarlas. No es que Lavoisier y sus colegas estuvieran ciegos a las anomalías. Por ejemplo, como Seymour Mauskopf (1988) relata con detalles esclarecedores[33], Lavoisier estaba muy interesado en la química de la pólvora e hizo algunos intentos diferentes para explicar su funcionamiento en términos de su teoría. El interés de Lavoisier por la pólvora no es una sorpresa después de todo, ya que fue comisionado de la Administración Real de la Pólvora desde 1775 y en esa capacidad estableció su residencia y laboratorio en el Arsenal de París. Estos intentos no tuvieron mucho éxito, y no menos que Claude-Louis Berthollet (1748-1822) utilizó el caso de la pólvora contra la teoría de la combustión de Lavoisier, citando precisamente la dificultad que Thomson informó más tarde[34]. El mismo Lavoisier nunca estuvo del todo satisfecho con la defensa inteligente pero torpe que pudo dar, aunque Berthollet se tranquilizó en este punto después de su "conversión" al sistema de Lavoisier por otras razones. Después de ese tropiezo en medio de París, la teoría de la combustión de Lavoisier avanzó y salió, inalterada. Thomson informó que Luigi Valentino Brugnatelli (1761–1818) había resuelto algunas de las dificultades con su concepto de "termoxígeno", que es oxígeno que se combina con otras sustancias conservando su luz y calorías[35]. Esto preocupó a Thomson como un movimiento descaradamente ad hoc, pero fue una forma de que la teoría lavoisieriana mantuviera su doctrina principal mientras abordaba una anomalía importante; tales ideas fueron entretenidas por los lavoisierianos cuando fue necesario, pero no fueron admitidos en el núcleo de su sistema.
La conclusión de Thomson sobre Lavoisier fue claramente negativa: en general, no se puede negar que la teoría de Lavoisier no ofrece una explicación suficiente de la combustión. Thomson no estaba solo en este tipo de juicio. Numerosos otros químicos que aceptaron que el oxígeno combinado con combustibles se mantuvieron escépticos sobre la explicación de Lavoisier sobre el calor y la luz en la combustión; estos "flogistonistas tardíos" a menudo mantenían un sistema en el que el oxígeno y el flogisto coexistían felizmente, a este último todavía se le asignaba la función de explicar lo que ahora identificaríamos como las relaciones energéticas en la combustión. Pero Lavoisier estaba más preocupado por mantener la simplicidad de su teoría que por ajustarse a todos los hechos conocidos. La historiografía de celebración posterior de la Revolución Química casi ha logrado borrar este aspecto de la historia de la memoria colectiva.
Además de la simplicidad y la integridad, también estaban en juego tipos más amplios de valores epistémicos. Una especie de conservadurismo epistémico fue uno de los valores defendidos por muchos flogistonistas, mientras que a los oxigenistas les cautivó la idea de reforma o novedad en sí misma. Hay un pasaje interesante de Cavendish que ilustra este punto: “será muy difícil determinar mediante el experimento cuál de estas opiniones es la más verdadera; pero como el principio comúnmente aceptado del flogisto explica todos los fenómenos, al menos tan bien como el del Sr. Lavoisier, me he adherido a eso[36] ".
El temperamento de Cavendish que se muestra aquí es sin duda un contraste informativo con el entusiasmo juvenil de Lavoisier que se declaró a sí mismo en 1773 que sus investigaciones estaban "destinadas a provocar una revolución en la física y la química", antes de que hubiera publicado incluso su primer ataque al flogisto.
Por otro lado, no puede ser que los principales flogistonistas se opusieran simplemente al cambio científico, ya que seguramente se deleitaban en hacer nuevos descubrimientos y también en elaborar algunas nuevas ideas teóricas. Muchos de los argumentos hechos en nombre del flogisto no fueron motivados por el conservadurismo sino por el pluralismo, en reacción contra el dogmatismo lavoisierianos. Esto es bastante contrario a la noción común de que los flogistonistas estaban cegados por el dogma. Declarando que la libre discusión debe ser siempre favorable a la causa de la verdad, recordó al lector el camino no dogmático que había recorrido en la ciencia:
Ninguna persona familiarizada con mis publicaciones filosóficas puede decir que parezco haber estado particularmente apegado a alguna hipótesis, ya que con frecuencia he confesado un cambio de opinión, y he expresado más de una vez una inclinación por la nueva teoría [lavoisieriana], especialmente esa misma parte importante de ella la descomposición del agua. (Priestley, 1796)
Priestley trazó un paralelo ominoso entre la política de la ciencia y la política más amplia que había puesto un final prematuro a la vida de Lavoisier en la guillotina en 1794, por su participación en el negocio de la recaudación de impuestos: “Como no quisieras ... reinado para parecerse al de Robespierre, pocos como somos los que permanecemos descontentos, esperamos que prefieras ganarnos con la persuasión que silenciarnos con el poder[37] ".
No creemos que todo esto fuera un autocontrol retrospectivo o la súplica rencorosa de un perdedor por sobrevivir. Priestley había expresado puntos de vista epistémicos similares incluso en el apogeo de su fama y éxito. Por ejemplo, en la carta de 1775 en la que anunció el descubrimiento de aire desflogistizado (oxígeno), Priestley escribió (1775):
“Es feliz cuando, con una fertilidad de invención suficiente para plantear hipótesis, una persona no es apta para adquirir un apego demasiado grande a ellas. Por este medio conducen al descubrimiento de nuevos hechos, y de un número suficiente de ellos resultará fácilmente la verdadera teoría de la naturaleza”.
Este pasaje siguió directamente su propuesta de que “el ácido nitroso es la base del aire común, y que el nitro se forma por una descomposición de la atmósfera”, a lo que agregó: "Pero puede que mañana piense lo contrario". Casi se puede escuchar un eco de Montaigne terminando sus pensamientos con "aunque no sé". En contraste, hubo un claro impulso absolutista en el lado oxigenista, quizás más atrozmente manifestado en la quema ceremonial del texto flogistonista de Stahl. Como lo describe Justus Liebig (1851), se trataba de "una fiesta en la que Madame Lavoisier, vestida de sacerdotisa, se entregaba a las llamas en un altar, mientras se cantaba un réquiem solemne, el sistema flogístico de la química[38]".
¿Qué vamos a concluir, habiendo visto estos argumentos de ambos lados? Nuestra sensación es que Lavoisier fracasó claramente en dar una descripción completa de la combustión. ¿Significa eso que su teoría estaba simplemente equivocada y, por lo tanto, su agradable simplicidad era inmaterial? O, para ser un poco más sutil: ¿no deberíamos decir, siguiendo el punto de vista de Bas van Fraassen sobre la evaluación de la teoría (1980), que la teoría de la combustión de Lavoisier no era empíricamente adecuada, y no tenía mucha simplicidad, que es sólo un virtud pragmática, ¿podría redimirla? El asunto no es tan sencillo, porque la adecuación empírica viene en partes y partes (en varias áreas de problemas tratadas por cada teoría), y además viene en grados en cada parte y parte[39]. La incomodidad de una estricta jerarquía de valores entre la adecuación empírica y las virtudes pragmáticas se hace evidente cuando nos preguntamos si una pequeña ventaja en la adecuación empírica no sería compensada por una gran cantidad de simplicidad adicional u otras virtudes.
El problema se agrava cuando reconocemos que la adecuación empírica en sí misma no es una variable de valor único en ningún caso; ¿Cómo sopesamos una ventaja en la adecuación empírica en un área con una desventaja en otra área? Todo lo que puedo decir, en esta etapa, es que no podemos decir nada definitivo acerca de la elección oxígeno-flogisto, al menos hasta que hayamos tenido una visión integral de todos los problemas relevantes y todos los valores epistémicos relevantes en la evaluación de las soluciones.
Donde los diferentes valores chocan entre sí, el epistemólogo se encuentra en un territorio incómodo. ¿Cómo podemos decir cuál de los valores apreciados por diferentes científicos históricos fue más valioso? ¿Tenemos algún derecho a emitir tales juicios? En mi opinión, esta no es una cuestión de derechos, ya que no vamos a hacer nada a los actores del pasado a nuestro juicio. Más bien, creemos que la pregunta es sobre el presente, y creo que tenemos el deber de hacer tales juicios con nosotros mismos. Como académicos de la ciencia, o como cualquier ciudadano responsable que considere cuestiones de conocimiento, necesitamos emitir juicios sobre la base de los valores epistémicos del presente. Entonces es imposible evitar aplicar estos juicios de alguna manera al pasado, al igual que es imposible mantener nuestros valores éticos presentes completamente fuera de nuestros estudios de historia en general. No es suficiente decir que no tenemos derecho a juzgar si un acto pasado, digamos un genocidio, fue correcto o incorrecto. Por el contrario, es importante permanecer alerta a los juicios que tenemos sobre los valores epistémicos que operan en la ciencia pasada, porque afectarán nuestros juicios sobre la ciencia actual. Lo que celebramos y condenamos en la ciencia pasada, aunque sea implícita o sutilmente, no puede permanecer separado de manera segura de cómo tratamos con la ciencia actual. Por tanto, no es irrelevante plantearse preguntas como si la simplicidad o la completitud fue la virtud superior en la elección del flogisto-oxígeno. Nuestra forma honesta y sesgada de plantear la pregunta sería: ¿qué actitud era (y es) más racional o científica, entre adaptar las teorías a los nuevos fenómenos de los que aprendemos y dar dominio dogmático a una teoría favorecida?
4.9 Instancias divergentes del mismo valor
Habiendo abordado los diferentes pesos dados a diferentes valores epistémicos por los lados opuestos en la Revolución Química, ahora debemos examinar una complicación adicional planteada por Kuhn (1977), que incluso un mismo valor epistémico puede ser interpretado y ejemplificado de formas divergentes, lo que lleva a conclusiones muy diferentes e incluso a acusaciones mutuas de traición al valor en cuestión.
Hay algunos casos significativos de instanciación de valores divergentes en la Revolución Química. Ambos lados valoraban la unidad, y cada lado citó el tipo de unidad que pudo lograr como evidencia persuasiva a su favor. Hubo cierta convergencia en esto, ya que ambos sistemas unificaron la combustión, la calcinación y la respiración de manera similar. Pero más allá de eso, hubo una divergencia significativa en lo que se unificó y cómo. La teoría calórica de Lavoisier unió las explicaciones de la combustión y los cambios de estado. Sus ideas sobre el oxígeno conectaban la combustión y la acidez, ya que muchos productos de la combustión eran ácidos. Por el lado del flogistonista, existía una agradable unidad teórica sobre el comportamiento de los metales: sus propiedades comunes, su calcinación/reducción y su reacción con los ácidos. La teoría del flogisto también conducía más a una gran unidad de todas las sustancias imponderables, como manifestaciones del "fuego elemental": flogisto, electricidad, luz, magnetismo, etc. (para las conexiones entre el flogisto y la electricidad).
De manera similar, pero aún más sorprendente, la adhesión de ambos lados al valor de la sistematización se manifiesta en cada lado acusando al otro de ser arbitrario y desordenado. Desde el lado flogistonista, la acusación contra los lavoisierianos fue que no se adhirieron a la regla de asignar causas similares a efectos similares. Tanto Priestley como Kirwan (1789) utilizaron este argumento en los debates constitucionales, para combatir la negativa oxigenista de reconocer la presencia común de flogisto en diversas sustancias. Por su parte, Lavoisier tenía un claro desdén por las continuas complicaciones y los cambios mutuamente conflictivos que varios flogistonistas introducían en sus teorías en sus intentos de afrontar los desafíos planteados por diversos fenómenos nuevos:
¿Por qué, entonces, necesitamos recurrir a un principio hipotético, cuya existencia se supone alguna vez y nunca se ha probado? que en un caso debe considerarse pesado, y en otro vacío, y al que, en algunos casos, es necesario suponer incluso un peso negativo; una sustancia que en algunos casos pasa a través de los vasos y en otros es retenida por ellos; un ser que sus mantenedores no se atreven a definir rigurosamente, porque su mérito y su conveniencia consisten incluso en la incertidumbre de las definiciones que se le dan?
Ahora, la declaración de Lavoisier es engañosa en la medida en que implica que cada flogistonista o cada versión de la teoría del flogisto sostuvo estas creencias mutuamente contradictorias sobre la naturaleza del flogisto; más bien, hubo diferentes versiones de la teoría del flogisto, en algunos casos sucesivas en el tiempo, que no coincidían en todos sus detalles. Aún así, Lavoisier estaba haciendo una demanda razonable de que el sistema del flogisto en su conjunto debería desarrollarse de una manera más sistemática. A diferencia de Lavoisier, ningún líder del lado flogistonista tuvo la voluntad o los medios para hacer que todos los de su lado cantaran la misma hoja de himno.
Un examen de los argumentos de ambos lados también revela su lealtad común a lo que llamaremos "empirismo": un compromiso para evitar invocar hipótesis extrañas, para permanecer cerca de los hechos observables y las ideas derivadas de esos hechos. En el pasaje citado anteriormente, Lavoisier y sus colegas denunciaron al flogisto como una entidad hipotética cuya existencia “nunca se supuso, nunca se demostró”. En su propia teoría, afirmaron, "no se admite nada más que verdades establecidas"; la suya era una doctrina que explica todos los hechos de la química sin ningún supuesto. La mayoría de los flogistonistas no fueron menos inflexibles sobre su empirismo. Ya hemos citado a Priestley diciendo que no tenía un fuerte apego a las hipótesis y que las consideraba principalmente como un medio para obtener nuevos hechos.
Habiendo revisado esas instancias de valor divergentes, la pregunta de nuestro propio juicio vuelve a surgir: ¿cuál creemos que era el tipo de unidad más valiosa, el tipo de sistematicidad más útil y el tipo de empirismo más genuino? En cada caso, no vemos ninguna forma irrefutable de argumentar que uno era más importante que el otro.
4.10 ¿Qué sucedió realmente en la revolución química?
Parece claro que cada uno de los sistemas oxigenista y flogistonista tenía sus propios méritos y dificultades, y que existían diferentes estándares según los cuales uno u otro estaba mejor apoyado por la evidencia empírica. En cierto modo, esto es solo una indicación de que el apoyo probatorio no es una cuestión sencilla de conexiones lógicas o probabilísticas entre la teoría y la observación, sino una relación compleja mediada por valores epistémicos, que pueden ser divergentes y contextuales. Nuestro propio juicio es que ambos sistemas fueron parcialmente exitosos en sus intentos de alcanzar metas valiosas, y que no había razón para favorecer claramente a uno sobre el otro.
Pero si no había una justificación clara para la elección del oxígeno sobre el flogisto, ¿por qué los químicos tomaron esa decisión? ¿Por qué ocurrió la Revolución Química? Hay una gran futilidad en la empresa de explicar por qué la gran mayoría de químicos se pasó rápidamente al lado de Lavoisier, porque en realidad eso no fue exactamente lo que sucedió.
Existe una extensa literatura sobre la Revolución Química, que no podremos examinar aquí de manera exhaustiva; en cambio, remito al lector a la revisión crítica actualizada y completa de esta literatura realizada por John McEvoy[40]. Nuestro propio objetivo aquí es hacer avanzar una tesis revisionista particular: la Revolución Química no consistió en una conversión rápida y casi universal de la comunidad química a la teoría de Lavoisier. En primer lugar, debemos resistirnos a dejarnos engañar por declaraciones triunfalistas que emanan de Lavoisier, de sus defensores contemporáneos y de sus glorificadores póstumos. Pero las declaraciones de una victoria limpia también se pueden encontrar en algunos lugares bastante inesperados. Tomemos, por ejemplo, la frase inicial de la defensa de Priestley de la teoría del flogisto de 1796: "Ha habido pocas, si es que ha habido alguna, revoluciones en la ciencia tan grandes, tan repentinas y tan generales como la prevalencia de lo que ahora se suele denominar el nuevo sistema de la química”, o la de los antiflogistas, sobre la doctrina de Stahl, que en un momento se pensó que había sido el mayor descubrimiento que jamás se había hecho en la ciencia. Quizás fue solo un lamento exagerado del perdedor, pero extrañamente la misma idea también se puede encontrar en las obras de algunos historiadores muy expertos, como Robert Siegfried: “De todas las revoluciones conocidas en la historia de la ciencia, la química es quizás la más dramática. . . . Solo 20 años separan las primeras exploraciones de Lavoisier de la química de los gases y la capitulación pública de Richard Kirwan, el último defensor europeo significativo de los puntos de vista flogísticos[41]". McEvoy señala: “la rapidez, brevedad y ritmo de la Revolución Química” como algunos de los factores clave que “la marcaron en la mente de muchos comentaristas como posiblemente el mejor ejemplo de una revolución clásica en la historia de Ciencias[42]”.
Sin embargo, una mirada razonablemente cercana a la literatura primaria desde alrededor de 1790 en adelante debería hacer evidente que hubo numerosos químicos que se negaron a subirse al carro de Lavoisier, a quienes llamaremos“anti-anti-flogistonistas”.
Aunque Lavoisier fue lo suficientemente optimista como para declarar en 1790 (en una carta a Benjamin Franklin) que ha tenido lugar una revolución en un área importante del conocimiento humano, sabía muy bien que todavía había muchos que no estaban convertidos, especialmente en Alemania y Bretaña; incluso los químicos franceses seguían “divididos”, como reconoció en la misma carta. Muchos de los antiflogistonistas eran químicos respetables y respetados, no ancianos impulsados ??por puro conservadurismo o dogmatismo. Hubo al menos tres tipos diferentes de disidentes en el período posterior a la publicación de Elements of Chemistr y de Lavoisier en 1789, que a menudo se ve como el punto en el que la Revolución Química se consolidó irreversiblemente. En primer lugar, hubo algunos intransigentes. Priestley encabeza esta lista, pero es solo una pequeña parte de la imagen. Scheele no sobrevivió el tiempo suficiente para que sus credenciales "acérrimas" fueran probadas verdaderamente, pero hasta su muerte en 1786 no mostró signos de renunciar a la teoría del flogisto. Una de las figuras más llamativas es Jean-André De Luc, cuya objeción se basó en su teoría de la lluvia que postula la transmutación del aire atmosférico en agua, así como en su disgusto general por la erupción y lo revolucionario en la ciencia y la política.
La segunda categoría de disidentes buscaba un compromiso o una neutralidad deliberada. Allchin, en su artículo titulado acertadamente "Phlogiston After Oxygen" (1992), presenta un caso convincente de que muchos químicos admitieron la existencia de oxígeno por consideraciones gravimétricas, mientras que mantuvieron el flogisto para lo que llamaríamos consideraciones energéticas[43].
De manera más general, la gente a menudo aceptaba la teoría de Lavoisier solo parcialmente, eligiendo y eligiendo lo que tenía sentido para ellos. El viejo flogistonista Pierre-Joseph Macquer estaba adoptando este tipo de enfoque cuando murió en 1784, e incluso el cercano colega y aliado de Lavoisier, Claude Louis Berthollet, se mantuvo escéptico sobre algunas de las ideas de Lavoisier, especialmente su teoría de los ácidos.
Hubo muchos otros que vieron claramente algún mérito en la química de Lavoisier pero no consideraron suficiente para llegar a un veredicto claro a favor de ella. Como se discutió anteriormente, Cavendish (1784) dio una visión lúcida de cómo ambas teorías podían explicar los fenómenos que observaba, al tiempo que expresaba su preferencia por quedarse con el flogisto.
Aún más interesante es la tercera categoría de disidentes, quienes reconocieron plenamente que el sistema de Lavoisier se había establecido pero también sintieron que su tiempo pasaba rápidamente. Muy sugerente a este respecto es el siguiente fragmento de conversación científica con el que me topé, del año 1800. William Herschel (1738-1822) acababa de detectar radiación infrarroja proveniente del sol, que vio como rayos calóricos separados de la luz. rayos por medio del prisma[44]. Joseph Banks (1743-1820) escribió para felicitar a Herschel por este descubrimiento trascendental, pero tenía un consejo: “Creo que todos mis amigos opinan que el sistema francés de química, en el que los nombres adoptados últimamente por sus químicos son fundada, ya se tambalea sobre su base y es probable que pronto sea subvertida. Por tanto, me atrevo a sugerirle si no será mejor para usted. . . utilizar el término Calor Radiante en lugar de Calórico; por el uso de esta última palabra debería parecer como si hubieras adoptado un sistema de química que probablemente nunca hayas examinado ".
Herschel aceptó felizmente el consejo de Banks: "Tengo el honor de su carta y estaré muy dispuesto a cambiar la palabra calórico por calor radiante, que expresa muy bien mi significado". Banks era un botánico y no un químico conocido, pero si el antiguo presidente de la Royal Society y "todos sus amigos" estaban prediciendo la inminente desaparición de la química francesa en 1800, entonces debe haber algo que nos hemos perdido en nuestra historiografía habitual. ¿Qué tenía exactamente Banks en mente? Es imposible estar seguro, pero podemos adivinar bastante bien. Ya he discutido las dificultades de la teoría de la acidez de Lavoisier, su teoría de la combustión y su teoría del calor. Algunos de los críticos de nueva generación de Lavoisier estaban ubicados en el Londres de Banks en ese momento, incluidos Humphry Davy y Count Rumford, además de viejos disidentes como Cavendish y De Luc.
Quizás el caso más interesante de la nueva generación de químicos anti-Lavoisier fue el de Humphry Davy (1778-1829). Davy era un niño de unos 10 años cuando se publicó Elements of Chemistry de Lavoisier y fue uno de los que crecieron con la ortodoxia de Lavoisier, pero llegó a rechazarla después de un examen más detenido. Se hizo un nombre no solo con el gas hilarante y la electroquímica, sino poniendo un clavo en el ataúd de la teoría de los ácidos de Lavoisier con su argumento de que el cloro era un elemento y que el ácido muriático (clorhídrico ácido) no contenía oxígeno, solo hidrógeno y cloro. Después de la aceptación del trabajo de Davy, la teoría de la acidez del oxígeno de Lavoisier cayó claramente en desgracia entre los químicos. Junto con Rumford, Young y Cavendish, Davy también fue uno de los que montaron serios desafíos a la teoría calórica de Lavoisieria del calor, cuyo dominio nunca fue total.
David Knight comenta (1978, 4): “había esperanzas y temores generalizados hasta al menos 1810 de que Davy la restaurara [la teoría del flogisto] y derrocara las doctrinas francesas”. Entre los que expresaron tal esperanza en forma impresa se encontraba Sir George Smith Gibbes, médico y profesor de química en Bath, que más tarde sería médico de la reina Charlotte; en 1809 Gibbes opinó que los descubrimientos de Davy habían confirmado que, después de todo, Lavoisier estaba equivocado. Otro que abrigaba tales expectativas fue el joven Michael Faraday (1791-1867), quien escribió en 1812, citando a Davy como una autoridad:
“Me gustaría que no se sorprendiera si la vieja teoría de Phlogiston fuera adoptada nuevamente como la verdadera[45]”.
Tomando todo eso en consideración, ¿en qué podemos decir que realmente consistió la Revolución Química? Todavía tenemos que admitir que un número considerable de químicos se "convirtió" completamente a la química de Lavoisier al menos durante un tiempo, y que logró un claro dominio en los libros de texto. Sin embargo, también debemos reconocer que hubo casos comunes de conversiones parciales o a medias, en muchos casos con retención de flogisto. Agreguemos a eso no solo los flogistonistas acérrimos, sino también la generación más joven de disidentes que realmente tuvieron su educación científica después de la victoria de Lavoisier. Una cosa muy interesante acerca de esas dos generaciones es que de hecho se superpusieron significativamente en el tiempo, la nueva generación apareció antes de que todos los intransigentes se hubieran rendido.
Phlogiston nunca fue revivido verdadera y exitosamente, y de hecho hay muchos sentidos en los que Lavoisier y sus colegas provocaron una "revolución" en la química, pero no fue un asunto repentino y claro. Fue una lucha multifacética que no terminó en un acuerdo unánime ni estableció una ortodoxia inmutable.
4.11 Pesos, composición y práctica química
Habiendo modificado la descripción de la manera en que el sistema oxigenista triunfó sobre el sistema flogistonista, volvemos ahora a la tarea de la explicación. Incluso en mi historia revisionista se da el caso de que una clara mayoría de químicos finalmente abandonó el flogisto, y ese hecho queda por explicar.
La importancia del peso
Hay algo de crucial importancia sobre la Revolución Química que aún no hemos considerado completamente. Muchos historiadores y químicos bien informados han argumentado que la contribución más importante de Lavoisier a la química fue su énfasis en el peso: su reconocimiento de su importancia y su uso de mediciones de precisión para rastrearlo a través de cambios químicos. Según este punto de vista, el oxígeno no era realmente el centro del sistema químico de Lavoisier, por mucho que el propio Lavoisier estuviera enamorado de él; más bien, fue un enfoque constante en los pesos lo que hizo que el nuevo sistema fuera superior al sistema flogistonista. Parece que lo que los oxigenistas consideraban el conjunto de argumentos más decisivo contra el sistema flogistonista se basaba en la consideración de los pesos en las reacciones químicas. Por el contrario, los flogistonistas no estaban tan preocupados por el peso, aunque algunos de ellos lo reconocieron como una propiedad relevante e incluso sabían cómo medirlo extremadamente bien[46].
¿De dónde vino esta diferencia y cómo afectó la diferencia en cuestiones relacionadas con el peso al estado probatorio de los sistemas en competencia? El enfoque en el peso no entró mucho en la discusión, porque no es realmente un valor epistémico. Un intento de descifrar qué tipo de cosa es, nos llevará a otra dimensión del razonamiento probatorio.
Uno puede tener una visión clara de qué lado tenía razón en la Revolución Química, si uno comparte la preocupación de Lavoisier por el peso y su concepción del peso como una cantidad conservada. Tome la descomposición y recomposición del agua. La opinión de Lavoisier es clara como el cristal: tomamos 100 g de agua y sacamos 15 g de hidrógeno y 85 g de oxígeno (aquí estoy usando las cifras del propio Lavoisier[47]); y luego, podemos juntar esas cantidades precisas de hidrógeno y oxígeno, y hacer 100 g de agua nuevamente. ¿Qué mejor prueba que esta podría tener uno para la idea de que el agua es un compuesto formado por hidrógeno y oxígeno? Por el contrario, considérese la historia del flogistonista: el hidrógeno está destinado a ser "agua flogística" y el oxígeno "agua desflogisticada". Cavendish y Priestley pensaron que el agua era la base de todos los gases, pero no dieron una buena historia sobre por qué la flogisticación debería hacer que el agua sea menos densa que la desflogisticación, o una medida precisa de cuánto flogistón entraba en la parte del agua que se convirtió en hidrógeno (o vino de la parte que se convirtió en oxígeno). Uno puede ver fácilmente la fuerza del relato de Lavoisier.
Sin embargo, no debemos apresurarnos en nuestro juicio. ¿Tenían los químicos durante la Revolución Química buenas razones para aceptar los argumentos basados ??en el peso de Lavoisier sobre la constitución de ciertas sustancias clave? Esta pregunta es crucial, porque los argumentos basados ??en el peso con respecto a la constitución proporcionaron quizás la única evidencia disponible en ese momento que podría haber obligado racionalmente a los flogistonistas a rendirse. Priestley y algunos otros rechazaron claramente los argumentos basados ??en el peso.
¿Por qué lo hicieron y se equivocaron al hacerlo? El razonamiento de Lavoisier dado anteriormente se basaba en dos supuestos muy importantes:
(a) El peso es una medida buena y adecuada de la cantidad de todas las sustancias químicas.
(b) Se conserva el peso.
Si se aceptaran estos supuestos, habría un caso convincente para el uso de ponderaciones como la principal fuente de evidencia para las ideas constitucionales. Pero, ¿hubo buena evidencia para respaldar los supuestos (a) y (b) mismos? Al considerar esa pregunta, debemos comenzar por perder el prejuicio de que es el peso, por supuesto, es importante enfocarse, y que las cosas ingrávidas no pertenecen a la buena química. La suposición (a) no se mantuvo universalmente incluso dentro de la propia química de Lavoisier, ya que las dos primeras en su lista de sustancias simples, ligeras y calóricas, no tenían peso. El supuesto (b) también es complicado. Contrariamente a las intuiciones comunes, nunca hubo una razón metafísica profunda por la que deba conservarse el peso (y de hecho, la E = mc2 proclama que no es así). Por supuesto, no se conservan todas las cantidades mensurables. Algunos resultan como la temperatura; los valores de temperatura no se pueden sumar entre sí de manera significativa y no se conservan.
En cualquier caso, Priestley y algunos otros no estaban dispuestos a aceptar la conservación de peso precisa que afirma Lavoisier en la descomposición y recomposición del agua. Esto fue particularmente problemático dado que se suponía que la fuerza del argumento de Lavoisier derivaba de la rigurosa precisión de sus experimentos. William Nicholson (1753-1815), que aceptó el sistema de Lavoisier, dio algunas razones detalladas para dudar de los grados de precisión que se alega en las medidas de peso de Lavoisier. Este fue un aspecto del trabajo de Lavoisier que lo enfadó y lo hizo dudar[48]:
Debo pedir permiso para observar que estas largas filas de figuras, que en algunos casos se extienden hasta mil veces la sutileza del experimento, sólo sirven para exhibir un desfile del que la verdadera ciencia no necesita: y, más que esto, que cuando el grado real de exactitud en los experimentos está oculto a nuestra contemplación, estamos de alguna manera dispuestos a dudar si la exactitud escrupulosa del uso de los experimentos es realmente tal que haga que las pruebas de l'ordre demostratif.
Para aquellos con conocimientos modernos, es casi imposible no simpatizar con Nicholson aquí, ante la absoluta convicción de Lavoisier de que 85:15 era la proporción correcta de oxígeno-hidrógeno en el agua, en lugar de algo como 8: 1.
Composicionismo versus Principialismo.
Recapitulemos el argumento dado hasta ahora: si uno acepta la primacía de la contabilidad de los pesos de Lavoisier, entonces hay evidencia empírica muy fuerte para el sistema oxigenista sobre el sistema flogistonista; pero ¿por qué debería uno aceptar la primacía de las consideraciones de peso? En el centro de esa pregunta está si se deben aceptar los supuestos (a) y (b) anteriores y por qué. Hemos visto que no son evidentes por sí mismos.
Tampoco fueron probados por evidencia empírica directa. Más bien, fueron asumidos por Lavoisier y sus seguidores, y utilizados como parte del tejido de las prácticas experimentales mediante las cuales produjeron evidencia empírica. El seguimiento de los cambios químicos por medio del peso era una actividad epistémica clave dentro del sistema oxigenista y se adaptaba bien a muchas de las otras prácticas de Lavoisier. Pero no encajaba bien en el sistema flogistonista, aunque no estaba del todo ausente allí. La aritmética de los pesos químicos surgió dentro de una tradición de conocimiento químico que se llamo composicionismo, que debe contrastarse con el principialismo subyacente a las doctrinas flogistonistas[49]. Cada sistema incorporó una doctrina metafísica significativa sobre la ontología fundamental de las sustancias químicas, que diferían marcadamente entre sí. Voy a argumentar que una tendencia creciente hacia el composicionismo creó un clima que fue en su mayor parte agradable al sistema oxigenista y claramente desfavorable para el sistema flogistonista. Creemos que este fue el factor más importante responsable de la victoria oxigenista. Al identificar las tradiciones composicionista y principialista, me baso en el trabajo bien establecido de muchos historiadores de la química, especialmente Siegfried, aunque la terminología exacta es mía[50]. Cuando decimos "tradiciones" arriba estamos usando el término en un sentido vago. Para ser más preciso dentro de un propio marco analítico. El sistema flogistonista fue una instancia particular del principialismo, y el sistema oxigenista fue una instancia particular del composicionismo.
Una actividad epistémica fundamental del tipo de sistema composicionista fue describir las sustancias químicas como elementos o compuestos formados por esos elementos. Además, existían las actividades más experimentales de descomponer compuestos en sus elementos y recomponerlos a partir de esos elementos. Cuando se podía hacer tanto la descomposición como la recomposición, se consideraba como la mejor prueba de la presunta composición de una sustancia. Estas prácticas requerían la presunción de que los componentes eran unidades estables que se conservan mediante reacciones químicas. Esa presunción también fundamentó la actividad de explicar las reacciones químicas como la reorganización de componentes básicos distintos y estables que conservan su identidad en todo momento incluso cuando sus propiedades no se manifiestan en un estado de combinación.
Para aquellos que han sido educados en la ciencia moderna, el composicionismo puede parecer un sentido común o una condición necesaria de la química misma. De modo que es útil saber qué alternativas había a la química composicionista, y el principal contendiente del siglo XVIII fue el principialismo. Este es un tipo de sistema formado alrededor del concepto de principios, es decir, sustancias fundamentales que imparten ciertas propiedades características a otras sustancias. Las actividades epistémicas definitorias en el principialismo fueron: clasificar sustancias de acuerdo con propiedades observables; explicar las propiedades de las sustancias por referencia a principios; y efectuar transformaciones de sustancias mediante la aplicación (o retirada) de principios. Al igual que el composicionismo, el principialismo era un tipo de sistema que tenía muchas instancias, que compartían estas tres actividades centrales. Es importante señalar que la ontología principialista suponía una asimetría entre los principios y las demás sustancias que son transformadas por ellos, siendo los principios activos y los otros pasivos. Hay algunos viejos ecos en el principialismo, incluida la vieja metafísica del sustrato de elementos.
O, de hecho, podemos rastrear el principialismo a través de la noción alquímica de la transmutación de sustancias; Priestley escribió una vez a Benjamin Franklin: "Sonreirás cuando te diga que no desespero en absoluto por la transmutación de los metales". Sin embargo, quizás el ejemplo de principialismo más vívido para los historiadores modernos de la ciencia sea el sistema flogistonista de la química, originado por Johann Becher (1635-1682) y Georg Ernst Stahl (1660 1734) y practicado hasta la época de Lavoisier, la mayoría famoso por Priestley.
No es que las operaciones químicas en sí mismas puedan clasificarse inequívocamente como transformación principialista o descomposición-recomposición; eso sería asumir que las prácticas experimentales eran completamente no teóricas. Muchas operaciones químicas eran fácilmente susceptibles de ambas interpretaciones; y esto no es una mera cuestión de interpretación de sillón, ya que las concepciones afectaron también a los experimentos reales. En este sentido, es interesante observar un debate temprano sobre la coherencia de la práctica de analizar sustancias mediante la aplicación de fuego: algunos tenían la confianza composicional de que el fuego solo produciría descomposiciones, mientras que otros mostraron preocupaciones principistas de que el fuego transformaría el sustancias que se analizan[51].
¿Cómo llegó a dominar el composicionismo? Varios historiadores de la química han escrito de manera instructiva sobre el origen del composicionismo, remontándolo a prácticas experimentales de descomposición-recomposición, así como a ideas sobre las “afinidades electivas” entre varias sustancias químicas. Como han explicado en detalle Siegfried, el origen de la química composicionista se remonta al menos a los filósofos mecánicos del siglo XVII, llegando a ser plenamente operativa a finales del siglo XVIII[52].
El sistema de química de Geoffroy se basaba firmemente en el marco composicionista, que representaba compuestos como combinaciones de partes indestructibles y reacciones químicas como reordenamiento de esas partes[53]. A eso Geoffroy agregó el concepto de afinidad y su fuerza relativa, lo que permitió explicar por qué ciertas combinaciones químicas se daban con preferencia a otras.
Creo que fue la adopción generalizada del composicionismo lo que dio supuestos (a) y (b) por encima de su apariencia de autoevidencia y, por lo tanto, llevó al triunfo de la química basada en el peso (Lavoisier). Y no es que el peso fuera el único parámetro posible para rastrear los componentes químicos, sino que fue el único que funcionó lo suficientemente bien para Lavoisier y sus contemporáneos. ¿Estaba el composicionismo en sí mismo apoyado por evidencia empírica suficiente? Esa es la pregunta incorrecta, como ya se señaló: supuestos como (a) y (b) permiten la producción de evidencia empírica; ellos mismos no son probados por la evidencia. Las formas de pensamiento de los principios y composicionistas se vincularon con diferentes prácticas experimentales. Más importante aún, el pensamiento principialista estaba vinculado con las prácticas de laboratorio de transformación; el pensamiento composicionista se vinculó con las prácticas de laboratorio de descomposición y recomposición. Cuando digo “vinculado”, lo que queremos decir es que los aspectos conceptuales y experimentales se refuerzan y moldean entre sí, en lugar de que uno provoque al otro de manera unidireccional; esto expresa lo que deseamos decir con la "coherencia" de un sistema de práctica. La química neumática de Priestley fue una práctica experimental fundamentalmente transformadora, como lo muestra claramente el examen de su principal tratado sobre el tema. Antes de comenzar a hacer los descubrimientos de todos los diferentes aires que lo hicieron famoso, estaba informando sobre cosas como "aire infectado con la respiración animal, o putrefacción" y "aire infectado con los vapores de carbón ardiendo" . Priestley no habría tenido ninguna idea en particular en ese momento de que ninguna sustancia se combinara con el aire en el proceso de respiración o combustión. Más bien, la respiración fue vista como una operación transformadora, y eso fue todo. Cuando relataba los trabajos anteriores que formaron el trasfondo de su propia investigación, Priestley ensalzó la virtud de Joseph Black, quien había descubierto que la entrada de aire fijo suavizaba las sustancias calcáreas.
Esta forma de pensar también combinó bien, al menos inicialmente, con la noción de Stahlia de que el flogisto era el principio que impartía inflamabilidad y metalicidad a las sustancias. A medida que procedía con sus propias investigaciones, Priestley adoptó firmemente esta forma principialista de pensar sobre el flogisto, hablando de manera rutinaria de cómo el aire se transformó mediante la adición de flogisto en una "gradación regular desde el aire desflogistizado, pasando por el aire común y el aire flogístico hasta aire nitroso ”.
Esta serie no fue una pieza ociosa de teorización, sino algo arraigado en sus diminutas operaciones de laboratorio. En sus numerosos experimentos que condujeron a (y se basaron en) estas concepciones, Priestley tomó nota de todo tipo de propiedades que se daban a las sustancias a medida que se modificaban (como el color, el olor, la elasticidad y la reactividad química con varias otras sustancias), pero el peso no era algo que notara con mucha frecuencia. Los cambios de peso parecían caprichosos en relación con la flogisticación, lo que significaba que el peso no se tomaba como una variable confiable con el propósito de extraer patrones estables en el comportamiento de la naturaleza. ¿Qué tipo de prácticas experimentales estaban relacionadas con el pensamiento de Lavoisier? También lo impulsaba la química neumática, pero de una manera muy diferente a Priestley. La fascinación de Lavoisier, desde su primer "año crucial", era cómo el aire era absorbido y distribuido por sustancias sólidas en determinadas reacciones químicas. Su pensamiento se basaba en la presunción de que las sustancias químicas tenían componentes: unidades estables que se conservan mediante reacciones químicas. Este enfoque en el rastreo de entrada y salida, combinado con la inclinación cuantitativa de Lavoisier, dio como resultado su método de "balance", o su inclinación "algebraica".
Habiendo entrado así en el modo de pensar composicionista, Lavoisier buscó y desarrolló experimentos conforme a la vieja práctica composicionista de descomposición y recomposición, que era el método más convincente para determinar las composiciones. En este contexto, es interesante examinar el trabajo experimental de Lavoisier sobre el calor, en el que colaboró ??con Pierre-Simon Laplace (1749-1827). Aunque Lavoisier y Laplace reconocieron que el calor no tenía un peso detectable (y en este artículo ni siquiera se comprometen fuertemente con la realidad material del calórico), está claro que asumieron que el calor es una cantidad conservada, y apuntaron a encontrar una medida cuantitativa de la misma para hacerla susceptible al pensamiento composicionista. Ellos inventaron el calorímetro de hielo para este propósito, detrás del cual estaba la noción química de Lavoisier de que el agua líquida era un compuesto de una cantidad definida de calorías (calor latente) por unidad de cantidad de hielo.
El calorímetro de hielo resultó ser difícil de usar en la práctica, y la práctica composicionista relacionada con el calor no resultó muy fructífera en la química de Lavoisier. El balance tuvo más éxito como instrumento de cuantificación. Permitió un seguimiento conveniente y precisó de los componentes químicos que tenían peso y fomentó un poderoso sistema de práctica composicionista. El saldo, por así decirlo, era el instrumento perfecto para gestionar el balance. Esta aritmética de pesos químicos fue la clave para que Lavoisier completara el esquema dado por el composicionismo.
En su contabilidad química de pesos, se estudiaron las reacciones químicas haciendo un seguimiento de los pesos de los ingredientes y productos, y la composición se determinó con la ayuda de la suposición de que si una sustancia se volvía más pesada después de una reacción, entonces debía haberse compuesto con otra sustancia. Este sistema composicionista basado en el peso tuvo su éxito sorprendente en la química neumática, donde, antes de Lavoisier, se había observado el papel químico de varios gases pero sus pesos no habían recibido suficiente atención. El éxito de la práctica composicional basada en el peso de Lavoisier reforzó la convicción de que el peso era la variable más importante en las reacciones químicas; sin embargo, a quienes no operaban en ese sistema de práctica les habría resultado difícil compartir esa convicción.
Para resumir: la clara ventaja probatoria del sistema oxigenista sobre la base de consideraciones de peso solo es válida si se acepta el composicionismo; los flogistonistas ignoraron los argumentos basados en el peso porque eran principistas. La Revolución Química tiene mucho más sentido cuando la vemos como una onda montada sobre una gran ola, que fue el establecimiento muy gradual del composicionismo. Es importante ver más allá del choque entre flogisto y oxígeno. Si queremos concebir la Revolución Química como el acontecimiento que dio origen a la "química moderna", debemos seguir a Robert Siegfried que la identifica como una revolución composicionista, cuyo punto final no fue Lavoisier, sino Dalton.
4.12 ¿De qué sirve el Phlogiston?
Habiendo refinado el relato descriptivo de la Revolución Química y considerado sus explicaciones, ahora estamos listo para abordar la pregunta normativa: ¿fue correcto que los químicos de finales del siglo XVIII y a principios del XIX rechazaran el sistema flogistónico de la química?
(1) ¿Hubo algún conocimiento que los científicos perdieron cuando rechazaron el sistema flogistonista?
(2) ¿Hubo algún conocimiento que pudiera provenir de la conservación del sistema flogistonista, cuyo desarrollo fue retrasado o impedido por su desaparición?
(3) ¿Hubo un efecto beneficioso de tener presentes tanto el flogisto como el oxigenista, en términos de lo que produjeron las interacciones entre ellos?
(4) ¿Habría habido más interacciones beneficiosas entre el sistema oxigenista y el sistema flogistonista, si se hubiera mantenido este último?
Las preguntas (1) y (3) se refieren a la actualidad histórica, y las preguntas (2) y (4) se refieren a la potencialidad. Las preguntas (1) y (2) tratan de los méritos del sistema flogistonista en sí mismo, en comparación con los méritos del sistema oxigenista en sí mismo; las preguntas (3) y (4) se refieren a los méritos de una forma pluralista interactiva de hacer ciencia en comparación con una forma monista. Además, cada una de las preguntas debe ser tratada en dos niveles diferentes: primero en relación con la competencia e interacción entre los sistemas flogisto y oxigenista, y segundo en el contexto más amplio de la competencia entre principialismo y composicionismo.
Beneficios de Phlogiston.
Hubo un área clara de "pérdida de Kuhn" en la Revolución Química, que ya hemos mencionado. Los flogistonistas explicaron las propiedades comunes de los metales diciendo que todos los metales eran ricos en flogisto; esta explicación se perdió con la Revolución Química, ya que no funciona si hacemos la conocida sustitución del flogisto por la ausencia de oxígeno (o, como decía Lavoisier, una fuerte afinidad por el oxígeno). Como dice Paul Hoyningen-Huene: solo después de más de 100 años se pudo recuperar el potencial explicativo de la teoría del flogisto en la química moderna. Había que esperar hasta el advenimiento de la teoría electrónica de los metales[54] .
No solo eso, sino que el relato del flogistonista en realidad tiene una estrecha resonancia con la noción moderna de que todos los metales comparten propiedades metálicas porque todos tienen un "mar" de electrones libres. Si fuéramos verdaderamente abiertos, reconoceríamos al flogisto como el precursor de los electrones libres. La conexión flogisto-electricidad no es en absoluto una fabricación retrospectiva de historiadores o filósofos. Personas que postularon una estrecha relación entre el flogisto y la electricidad en el siglo XVIII. Hubo algunas buenas motivaciones para esta identificación, de encontrar una gran unidad entre todos los fluidos imponderables): por ejemplo, se encontró que la electricidad podía ser utilizada para reducir los cálices a metales, que era un papel desempeñado por el flogisto. Por tales razones, el químico inglés John Elliott (1780) incluso propuso que el flogisto debería ser rebautizado como “electrón”. Más tarde, cuando la electrólisis del agua en 1800 terminó en un rompecabezas sobre por qué los gases de oxígeno e hidrógeno se producían en lugares separados, la respuesta de Johann Wilhelm Ritter fue que el gas hidrógeno era un compuesto de agua y electricidad negativa, y el oxígeno un compuesto de agua y electricidad positiva; esto se alineó exactamente con la noción anterior de Cavendish de que el hidrógeno era agua flogística, al hacer la identificación de flogisto con electricidad negativa).
Para mostrar que no son solo los filósofos de la ciencia locos o los científicos completamente pasados ??los que han tenido estos pensamientos locos sobre el flogisto, nos referimos al gran químico estadounidense Gilbert Newton Lewis (de la "regla del octeto" y la definición aún actual de acidez). En sus conferencias Silliman en la Universidad de Yale, Lewis (1926) declaró que los flogistonistas dieron "el siguiente gran paso en la clasificación química" después del trabajo de Boyle, "a través de un estudio del fenómeno que conocemos como reducción y oxidación, pero que primero se llamó flogisticación y desflogisticación[55] ”.
Lewis pensó que la desaparición del flogisto en la Revolución Química constituía una gran oportunidad perdida para la química: si ellos [los flogistonistas] solo hubieran pensado en decir “la sustancia que se quema cede su flogisto al oxígeno del aire y luego se combina con él, la teoría del flogisto nunca habría caído en descrédito. De hecho, es curioso observar ahora que no solo su nueva clasificación pero incluso su mecanismo era esencialmente correcto. Solo en los últimos años nos hemos dado cuenta de que todo proceso que llamamos reducción u oxidación es la ganancia o pérdida de una sustancia casi imponderable, a la que no llamamos flogisto sino electrones.
La declaración de Lewis aquí es un buen recordatorio de que el significado moderno de "oxidación" no tiene nada que ver inherentemente con el oxígeno; en esta área de la química, podría haber sido más sensato quedarse con la estructura conceptual básica detrás de la terminología de flogisticación/desflogisticación (ganancia/pérdida de electrones), en lugar de la terminología bastante confusa de reducción/oxidación. En otras palabras, si se hubiera mantenido la idea de flogisto, habría sido más fácil para los químicos y físicos enfrentarse a esa misteriosa sustancia que juega un papel crucial en las transformaciones químicas, que abunda en metales, entre otros lugares. Este potencial se sugiere claramente en el informe de Allchin (1997) sobre el éxito en el uso del concepto de flogisto en la enseñanza de las reacciones redox a los estudiantes de hoy en día[56].
Si nuestros pensamientos sobre las oportunidades teóricas perdidas parecen demasiado especulativos, al menos podemos admitir que la preservación del concepto de flogisto habría estimulado algunas investigaciones experimentales que no fueron abordadas bajo el paraguas de Lavoisieria. Si el flogisto hubiera sobrevivido y se hubiera mantenido su asociación con la electricidad, estamos seguro de que los científicos del siglo XIX habrían intentado aislar el fluido eléctrico de las sustancias ricas en flogisto, como los metales, utilizando cualquier medio plausible a su disposición. ¿No se le habría ocurrido a alguien golpear la superficie de un metal con poderosos rayos ultravioleta (ya descubiertos en 1802) en un intento de desacoplar el flogisto? Tan pronto como hubiera electrómetros lo suficientemente sensibles, se habría detectado el efecto fotoeléctrico. ¿Qué hay de intentar hacer pasar una corriente eléctrica entre dos electrodos a través de un vacío cercano, un tipo de cosa muy familiar de la práctica tradicional de extraer chispas de la electricidad estática? De hecho, Davy, y también Jöns Jakob Berzelius, tenían el conocimiento experimental de que la electricidad podía pasar a través del vacío. Davy incluso evaluó que el paso de la electricidad era más fácil en el vacío que en el aire, y para Berzelius todo esto era una prueba de la materialidad de la electricidad. Pero la descarga eléctrica en gases enrarecidos, a pesar de sus llamativas manifestaciones visuales, no recibió una atención científica seria y generalizada hasta la década de 1870, cuando Eugen Goldstein identificó los “rayos catódicos” como tales. ¿Es demasiado irresponsable especular que los rayos catódicos se habrían descubierto e investigado muy pronto si se hubiera fomentado aún más la línea de investigación flogisto-electricidad?
A Elliott le hubiera gustado el aislamiento experimental del "electrón". También podríamos decir que la teoría del flogisto dio una buena explicación de la producción de llama en combustión. Davy estaba descontento por el descuido de la luz en la teoría de Lavoisier, y escribió en su artículo juvenil publicado en 1799 que había dos defectos de la teoría de Lavoisier, a saber, la suposición de calórico material y “el descuido total de luz". ¿Pero el flogisto lo hizo mejor aquí? En la teoría moderna, la llama es un plasma, que es principalmente una mezcla de iones positivos y electrones. Como acabamos de mencionar, en una comprensión blanda de la teoría flogistonista de los metales, existe una razón clara para identificar el flogisto con los electrones libres. Eso encaja muy bien aquí, si tomamos la liberación de la llama como resultado de la disociación del flogisto (electrones) de la sustancia combustible. A principios del siglo XIX, William Brande (1814) en la Royal Institution llevó a cabo experimentos que mostraban que la llama estaba sujeta a atracción electrostática, pero este trabajo no logró abrir una nueva línea de investigación; el progreso se habría facilitado mucho mejor en un marco flogistonista-electronista. La explicación del flogistonista, por supuesto, necesita una modificación aquí, ya que tendía a tratar la llama solo como flogisto en lugar de flogisto mezclado con material deflogisticado y tampoco explicaba específicamente por qué el plasma debería brillar, pero aún así era menos desesperante que la historia lavoisieriana de que la llama era una mezcla de dos sustancias químicas imponderables, calorique y lumière, ambas desconectadas del oxígeno gaseoso.
Si se hubiera conservado el flogisto, también habría servido como recordatorio de que las reacciones químicas eran más que la agrupación y reagrupación de bloques de construcción gravimétricos. Whiggly hablando, el flogisto sirvió como una expresión de la energía potencial química, que el composicionismo basado en el peso del sistema oxigenista perdió por completo de vista. La tradición de Lavoisieria era bastante inestable en este aspecto. Por ejemplo, Lavoisier sembró la semilla de la destrucción de su propia teoría de la combustión, poniendo tanto énfasis en el peso y luego sin asignar peso a las calorías. De hecho, la teoría de la combustión de Lavoisier nunca llegó muy lejos al explicar la liberación de calor y luz en la combustión, sin el concepto de energía disponible. Para empezar a pensar en la energía, los químicos no deberían haber tenido que esperar la ayuda de personas como Mayer, Joule y Helmholtz y cosas como el vínculo entre el calor y el trabajo mecánico. Si el flogisto hubiera vivido, les habría dado a los químicos un extremo abierto productivo para comenzar a pensar en algo como la energía.
Este movimiento, ya será recordado de alguna manera o recientemente reinventado, no pasó desapercibido para algunos de los químicos victorianos. En la novena edición de la Encyclopaedia Britannica (1876), F. H. Butler identificó el flogisto como otro nombre para la energía potencial: “La supuesta sustracción de flogisto en la calcinación de metales. . . era todavía una pérdida de energía potencial, en virtud de la combinación del metal con el gas [oxígeno]; y la ganancia de flogisto fue un aumento de la energía potencial, asociado a la eliminación de oxígeno ". Butler reconoció esta noción como un avance distintivo hecho en el período flogistonista[57]: fue solo en la última parte del siglo XVIII cuando esa nada aireada se volvió comúnmente considerada como un componente íntimo y necesario de varios cuerpos sólidos y fluidos.
Según Odling (1871), la principal idea de los flogistonistas fue que los cuerpos combustibles poseen en común un poder o energía capaz de ser provocada y utilizada", y que "la energía perteneciente a los cuerpos combustibles es la misma en todos ellos, y susceptibles de ser trasladados del cuerpo combustible que lo tiene a un cuerpo incombustible que no lo tiene.
Lavoisier se había equivocado al ubicar la energía en el oxígeno gaseoso en forma de calórico, sin una explicación convincente de por qué el calórico contenido en otros gases no tendría la capacidad de causar combustión. Odling pensaba que los Stahlians, aunque ignoraban mucho de lo que se ha conocido desde entonces, eran conscientes de mucho de lo que luego se olvidaba. También citó a Alexander Rum-Brown (1866), otro destacado químico victoriano, por tener la misma opinión de que "no puede haber ninguna duda" de que la energía potencial era lo que los primeros químicos "querían decir cuando hablaban de flogisto”. De hecho, Odling y Crum-Brown habían sido anticipados por Liebig dos décadas antes (1851, 49-50), antes del uso generalizado del concepto de energía en química: muchos químicos, incluso en la actualidad, encuentran imposible hacer sin ciertos nombres colectivos, análogos a la palabra flogisto, para procesos que consideran pertenecientes a la misma clase o determinados por la misma causa. Pero . . . emplean, desde la época de Berthollet, términos que designan lo que se llama fuerzas.
Que Stahl y sus seguidores consideraran el flogisto como una sustancia material, si lo consideraran así, no debería interferir más con nuestro reconocimiento del mérito debido a su doctrina, que la circunstancia de Black y Lavoisier respecto al calórico como sustancia material, si lo consideráramos así, debería interferir con nuestro reconocimiento del mérito debido a la doctrina del calor latente.
Aunque el flogisto claramente no era exactamente energía potencial química como se entendía en 1871, Odling argumentó que los flogistas tenían, en su tiempo, posesión de una verdad real en la naturaleza que, perdida por completo en el período intermedio, ha desde que cristalizó en una forma definida. La doctrina de la energía, fue por supuesto "la generalización más grandiosa de la ciencia que jamás se haya establecido".
Al consideramos todo, creemos que está bastante claro que acabar con el flogisto tuvo dos efectos adversos: uno fue descartar ciertos problemas y soluciones científicos valiosos; el otro era cerrar ciertas vías teóricas y experimentales para el trabajo científico futuro. Quizás todo esté bien desde donde nos sentamos, ya que creo que el potencial frustrado del sistema flogistonista finalmente se realizó completamente, por algunas rutas muy tortuosas. Pero me parece bastante claro que la muerte prematura del flogisto retrasó el progreso científico de formas bastante tangibles. Si se hubiera dejado desarrollar, creemos que el concepto de flogisto se habría dividido en dos. Por un lado, a principios del siglo XIX alguien podría haber acertado con la conservación de la energía, desconcertado por esta entidad imponderable que parecía tener una especie de realidad esquiva que podía pasar de una sustancia ponderable a otra.
En ese universo paralelo, estaríamos hablando de la conservación del flogisto, y cómo el flogisto resultó tener todo tipo de formas diferentes, pero todas interconvertibles entre sí. Esto no sería más incómodo que lo que tenemos en nuestro universo actual, en el que todavía hablamos sobre el papel del "oxígeno" (generador de ácido) en el apoyo a la combustión y el número de iones de "oxidación". Por otro lado, el concepto de flogisto podría haber llevado a un estudio de los electrones sin pasar por una teoría atómica tan categórica y simplificada como la de Dalton. Los químicos podrían haber pasado directamente del flogisto a las partículas elementales, o al menos haber encontrado un camino alternativo de desarrollo que no pasara por la falsa simplicidad de la jerarquía átomo-molécula-materia en masa. Mantener la teoría del flogisto habría llevado a los químicos a prestar más atención al "cuarto estado de la materia", comenzando con las llamas, y habría servido como recordatorio de que la durabilidad del compositor Mantener vivo el flogisto podría haber desafiado la sencilla suposición daltoniana de que los átomos químicos eran físicamente unidades irrompibles. La supervivencia del flogisto en el siglo XIX habría sostenido una vigorosa tradición alternativa en química y física, que habría permitido a los científicos reconocer con más facilidad la maravillosa fluidez de la materia y comprender antes la naturaleza de los iones, las soluciones, metales, plasmas, rayos catódicos y quizás incluso radiactividad.
Si la interacción entre el flogisto y el oxígeno se hubiera mantenido, nos parece que los químicos habrían podido mantener una tradición híbrida muy dinámica a lo largo del siglo XIX. Una tensión productiva entre el principialismo y el composicionismo habría ayudado a mantener un pluralismo saludable en la química. A medida que el composicionismo se desarrolló en una forma más pura y se volvió cada vez más dominante en su marcha desde Lavoisier a Dalton y más allá, surgió la tentación de pensar en los fundamentos de la química como una cuestión de elección absoluta: suscribirse a un atomismo simplista o renunciar a cualquier discurso ontológico sobre sustancias químicas. Esto es lo que algunos comentaristas han recogido como la oposición del siglo XIX entre atomismo y positivismo. Si los químicos se hubieran resistido a tales dicotomías y hubieran permanecido más conscientes de los éxitos flogisto-principialistas mientras se aferraban al composicionismo básico, habrían podido desarrollar una visión más flexible de los "elementos" y una visión más matizada de los una incorporación más fácil de la electricidad y la termodinámica a la química. Por el contrario, también hay algo que llamó "efecto laguna". Hay muchas cosas que el sistema flogistonista no logró explicar satisfactoriamente, pero el sistema oxigenista ni siquiera trató de explicar. En tales casos, un beneficio de mantener el sistema flogistonista habría sido servir como recordatorio de los problemas no resueltos.
Nos gustaría cerrar con un llamado a la imaginación, sobre lo que podría constituir un nuevo conocimiento científico y de dónde podría provenir. Tenemos serias intenciones en nuestra discusión sobre el sistema flogistonista de la química, sobre sus méritos olvidados y los beneficios que podría haber traído al siglo XIX. Sin embargo, incluso si cada una de nuestras ideas específicas sobre el futuro perdido del flogisto resultara inútil, todavía tendría la esperanza de un efecto más amplio de seguir nuestras cavilaciones: la liberación de la imaginación científica del lector.
4.13 Propiedades únicas del agua
A lo largo de toda la historia de la existencia de nuestro planeta, el agua ha afectado a todos los aspectos del planeta. Es este material de construcción principal, y el medio ambiente que lo contiene, lo que asegura la propagación y evolución de la vida en la Tierra. Durante muchos siglos, los científicos han estudiado esta sustancia química, compuesta por la fórmula simple, H2O, y conocida como una de las moléculas más pequeñas y ligeras. A pesar de su simplicidad y tamaño, el agua de alguna manera juega el papel principal en todos los procesos biológicos y se considera "la matriz de la vida". La gran capacidad calorífica, la alta conducción de calor y la enorme cantidad de agua en los organismos contribuyen a la regulación de la temperatura y previenen las fluctuaciones de temperatura local, lo que nos permite controlar más fácilmente nuestra temperatura corporal. El alto calor latente de evaporación asegura la resistencia a la deshidratación y un alto enfriamiento en la evaporación. El agua, debido a la polaridad de sus moléculas, su alta constante dieléctrica y sus moléculas de pequeño tamaño, es un buen solvente principalmente para compuestos iónicos polares y sales. Posee propiedades únicas de hidratación con respecto a las macromoléculas biológicas (especialmente a las proteínas y ácidos nucleicos), que determinan sus estructuras tridimensionales y, en consecuencia, funciones en la solución. Esta hidratación hace que se formen geles, que pueden ser sometidos reversiblemente a la transición solución-gel que se encuentra detrás de muchos mecanismos celulares.
El agua se ioniza y asegura un flujo ligero de intercambio de protones y, por lo tanto, conduce a la variedad de interacciones iónicas en biología. “El agua es increíblemente multifacética, ningún otro líquido tiene esta propiedad”, dice F. Gaiger, un destacado experto en agua de la Universidad de Dortmund. Cree que “… el mundo de los líquidos se ha dividido, hay una serie de cuestiones fundamentales sin respuestas concluyentes. ¿Por qué esta sustancia se comporta, en muchos aspectos, de manera atípica? ¿Por qué el agua en su estado cristalino, es decir, en forma de hielo, tiene tantas “caras” —15[58]? El complicado comportamiento de esta sustancia está determinado por la estructura de sus moléculas (Fig. 2.1). En la molécula de agua, dos átomos de hidrógeno y un átomo de oxígeno están dispuestos en un ángulo de 104,3° entre sí. Debido a su polaridad eléctrica, ambas moléculas de agua pueden crear un vínculo especial entre ellas; las cargas parciales positivas en los átomos de hidrógeno y las cargas negativas en los átomos de oxígeno se atraen entre sí, formando puentes de hidrógeno. Este vínculo es 20 veces más débil que el que conecta los átomos de hidrógeno y oxígeno dentro de una molécula, pero excede la fuerza de atracción normal entre las moléculas, la fuerza de Van der Waals, en 60 veces. La presencia del enlace de hidrógeno en el agua es necesaria, pero es una condición insuficiente para explicar sus principales propiedades. Si bien, el enlace de hidrógeno es un componente necesario de la molécula de agua, por sí solo no representa las principales propiedades del agua. Esto solo se puede hacer cuando la estructura del agua se considera un sistema único[59].
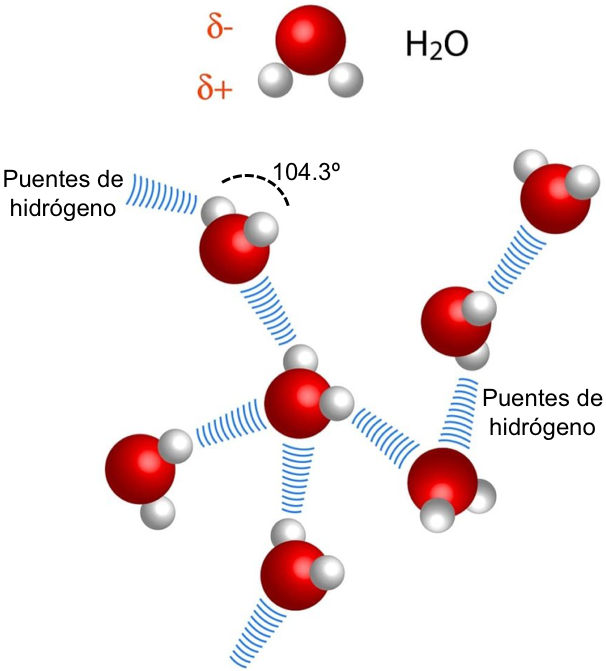
Fig. 4.1 Formación de enlaces de hidrógeno
Brian Greene refiere al agua en su libro “Until the end of time. Mind, matter and our search for meaning in an evolving universe[60]” como algo de lo más importante dado en el universo:
El agua es un caso de especial importancia. El oxígeno contiene ocho electrones, dos en la primera fila y seis en la segunda. Así pues, se muere de ganas por conseguir dos electrones más para llenar la segunda fila con el máximo que permite, que son ocho. El hidrógeno es una fuente abundante donde conseguir esos electrones. Cada átomo de hidrógeno contiene un solo electrón, que se sienta solo, cruzado de brazos, en la primera fila. Si un átomo de hidrógeno ve la ocasión de completar esa fila con un electrón más, lo hará de buen gusto. Así que el hidrógeno y el oxígeno llegan a un acuerdo para compartir un par de electrones, satisfaciendo así del todo al hidrógeno y llevando al oxígeno a solo un electrón de la felicidad orbital. Si incluimos un segundo átomo de hidrógeno que comparte del mismo modo un par de electrones con el oxígeno, la dicha es absoluta. Esos electrones compartidos unen el átomo de oxígeno con los dos átomos de hidrógeno, dando lugar así a una molécula de agua, H2O. La geometría de esta unión tiene implicaciones de largo alcance. Los empujes y tirones entre átomos llevan a todas las moléculas de agua a adoptar una forma de «V», con el oxígeno en el vértice y cada uno de los hidrógenos colgando de una de las puntas de la letra. El H2O no tiene carga eléctrica, pero el oxígeno es tan maniático con el llenado de sus capas orbitales que acapara los electrones compartidos, con el resultado de que la carga eléctrica de la molécula queda distribuida de forma irregular. El vértice de la molécula, donde se encuentra el oxígeno, queda con carga neta negativa, mientras que las dos puntas superiores, donde están los hidrógenos, quedan con carga neta positiva. La distribución de la carga eléctrica en la molécula de agua puede parecer un detalle esotérico. Pero no lo es. Resulta ser esencial para el origen de la vida. Gracias a la distribución sesgada de su carga eléctrica, el agua puede disolver casi todo. Los vértices de carga negativa, donde está el oxígeno, se agarran a todo lo que tengan la más mínima carga positiva, mientras que las puntas de hidrógeno, de carga positiva, se agarran a todo lo que tenga la más ligera carga negativa. En tándem, los dos extremos de la molécula de agua actúan como unas garras cargadas eléctricamente que mantienen separado casi todo lo que se sumerja en agua el tiempo suficiente.
La sal de mesa es el ejemplo más familiar. Compuesta de un átomo de sodio unido a un átomo de cloro, una molécula de sal de mesa posee una ligera carga positiva cerca del sodio (que dona un electrón al cloro) y una carga ligeramente negativa cerca del cloro (que acepta un electrón del sodio). Si tiramos sal en el agua, la parte del oxígeno del H2O (que tiene carga negativa) agarra el sodio (de carga positiva), mientras que el lado del hidrógeno del H2O (que tiene carga positiva) agarra el cloro (de carga negativa), desgarrando las moléculas de la sal y disolviéndolas en una solución. Y lo que es cierto para la sal también lo es para muchas otras sustancias. Los detalles varían, pero la distribución asimétrica de la carga en el agua hace que esta sea un magnífico solvente. Cuando nos lavamos las manos, aunque sea sin jabón, la polaridad eléctrica del agua trabaja a destajo para disolver cualquier sustancia extraña y llevársela con ella. Más allá de su utilidad para la higiene personal, la capacidad del agua para agarrar e ingerir sustancias es indispensable para la vida. El interior de las células es como un laboratorio químico en miniatura que para funcionar necesita mover con rapidez una gran colección de ingredientes: nutrientes que entran, desechos que salen, compuestos químicos que deben mezclarse para sintetizar sustancias necesarias para el funcionamiento de la célula, y tantas otras cosas. El agua hace que todo eso sea posible. Constituye alrededor del 70 % de la masa de la célula, y es para la vida el fluido de transporte. El premio Nobel Albert Szent-Györgyi lo resumió con elocuencia[61]:
El agua es la materia y la matriz de la vida, la madre y el medio. No hay vida sin agua. La vida solo consiguió salir del océano cuando aprendió a ponerse una piel, una bolsa en la que llevar agua. Todavía vivimos en el agua, solo que ahora la llevamos dentro.
Como poesía, es una elegante oda al agua y la vida. Como ciencia, todavía no podemos establecer la validez universal de esta aserción, pero tampoco conocemos ninguna forma de vida que ponga en entredicho la necesidad de agua.
Agua líquida En términos estadísticos, las partículas de agua en estado líquido forman tres puentes de hidrógeno y medio con sus vecinas. Estos puentes colapsan y se alinean nuevamente con una periodicidad de una milmillonésima parte de una fracción de milisegundo. En 1916, se propusieron nuevas nociones sobre la estructura de un líquido. Las primeras investigaciones estructurales de rayos X del agua se llevaron a cabo en 1922 por los científicos holandeses W. Keese y J. de Smedt. Demostraron que el agua líquida se caracteriza por la disposición ordenada de las moléculas de agua, es decir, el agua tiene una cierta estructura regular. La estructura del agua en un organismo vivo se parece en muchos aspectos a la estructura cristalina del hielo (Fig. 4.2).
 19.55.09.png)
Fig. 4.2 Estructura cristalina del hielo.
La “adhesividad” especial de una molécula es la razón por la que el H2O permanece líquido a una temperatura extrema de 100 °C. Los materiales químicamente afines en este sentido pasan por calor a un máximo de 25 °C. Cada molécula de agua en la estructura cristalina del hielo participa en la formación de cuatro enlaces de hidrógeno dirigidos a los vértices del tetraedro (Fig. 4.3). El centro tetraédrico contiene un átomo de oxígeno y dos vértices tienen cada uno un átomo de hidrógeno cuyos electrones están involucrados en la formación de un enlace covalente con el átomo de oxígeno. Los dos vértices restantes están ocupados por pares de electrones de valencia del átomo de oxígeno, que no participan en la formación de enlaces intramoleculares. En la interacción del protón de una molécula con un par solitario de electrones de oxígeno de la otra molécula, un enlace de hidrógeno parece más débil que el enlace intermolecular, pero lo suficientemente poderoso como para mantener unidas las moléculas de agua vecinas.
 20.00.09.png)
Fig. 4.3 Estructura tetraédrica de moléculas de agua.
Cada molécula puede formar simultáneamente cuatro enlaces de hidrógeno con otras moléculas en ángulos estrictamente definidos, lo que equivale a 109 ° 28 ′. Estos enlaces se dirigen a los vértices del tetraédrico, lo que no les permite al congelarse formar una estructura densa. La primera teoría sobre la estructura del agua fue propuesta por los investigadores ingleses J. Bernal y R. Fowler. Crearon un concepto sobre la estructura tetraédrica del agua y definieron el papel de los enlaces de hidrógeno en el agua. Se encontró que existen enlaces covalentes y de hidrógeno en el agua. Los enlaces covalentes no se rompen en las transiciones de fase del agua: agua-vapor-hielo. Solo la electrólisis, un proceso de calentamiento de agua sobre hierro y procesos similares, rompen los enlaces de covalencia del agua. Los enlaces de hidrógeno son 24 veces más débiles que los enlaces de covalencia. Al derretir el hielo y la nieve, los enlaces de hidrógeno permanecieron parcialmente en el agua y, en estado de vapor, se rompen todos (Fig. 4.4).
 20.01.49.png)
Fig. 4.4 Esquema de destrucción parcial y formación de enlaces de hidrógeno en la descongelación del hielo (el tiempo de salto es de 10 a 12s).
Posteriormente, se desarrolló una idea según la cual se propuso el agua líquida como un pseudocristal (Fig. 4.5); en él, se dice que las moléculas tetraédricas individuales de H2O están unidas entre sí por enlaces de hidrógeno direccionales, formando estructuras hexagonales como en la estructura del hielo. Hielo Ih En la abreviatura Ih, la letra h denota hexagonal. Este tipo de hielo está flotando en mares y océanos; es uno de los 15 tipos de estados cristalinos del agua que se conocen hoy en día, y esta forma se encuentra en la Tierra con más frecuencia que otras. Las moléculas están dispuestas de tal manera que cada elemento tiene 4 vecinos a los que están unidos por puentes de hidrógeno. Como resultado, una red de base hexagonal está formada por átomos de oxígeno.
Fig. 4.5 Agua como pseudocristal
La investigación cristalográfica de rayos X realizada por J.P. Morgan y B.E. Warren demostró que una estructura como la del hielo es inherente al agua[62]. En el agua, como en el hielo, cada átomo de oxígeno está rodeado por átomos de oxígeno como en el tetraedro. La distancia entre moléculas vecinas no es idéntica. A 25 °C, cada molécula de agua en el marco tiene un vecino a una distancia de 0.277 nm y tres a una distancia de 0.294, en promedio 0.290 nm. La distancia media entre los vecinos más cercanos de la molécula es aproximadamente un 5,5% mayor que entre las moléculas de hielo. Las otras moléculas están a distancias intermedias entre la primera y la segunda distancias vecinas[63]. La distancia 0.41 nm es una distancia entre los átomos O– H en una molécula de H2O.
Las principales diferencias entre la estructura del agua líquida y la estructura del hielo son una disposición más difusa de los átomos en la red y una alteración del orden remoto. Las fluctuaciones de calor dan como resultado la flexión y la rotura de los enlaces de hidrógeno. Las moléculas de agua de las posiciones de equilibrio entran en los vacíos vecinos de la estructura y permanecen allí durante algún tiempo, ya que estos vacíos tienen los correspondientes mínimos relativos de energía potencial. Esto conduce a un aumento del número de coordinación y a la formación de los defectos de la red, cuya presencia determina las propiedades anómalas del agua. El número de coordinación de moléculas (el número de vecinos más cercanos) varía de 4,4 a 1,5 ° C a 4,9 a 83 °C.
Hielo-? bajo la presión del agua, aumentada 600.000 veces, las moléculas de agua se enganchan tan fuertemente entre sí que las diferencias entre los puentes de hidrógeno y los enlaces químicos convencionales desaparecen por completo. Ice-X posee la mayor densidad entre las formas cristalinas conocidas de H2O (2,51 g/cm3). Su presión permanece fuerte incluso a una temperatura de 500 °C[64].
En estado gaseoso, las moléculas de agua se separan y se embarcan en un viaje en solitario. Solo ocasionalmente sus elementos individuales se unen entre sí sobre la marcha. Al transformarse en vapor, el agua absorbe una enorme cantidad de energía: 2258 J/g (en comparación, con el etanol es sólo 854 J/g). Durante la condensación, esta energía se libera nuevamente. Por eso el vapor de agua puede provocar quemaduras graves. ¿Cuándo y en qué se transforma el agua? Para tener una imagen más gráfica, estos datos se presentan de la siguiente forma: casi todas las escalas de temperatura tienen que ver con el agua, incluida la escala de temperatura de Kelvin, que se basa en el punto triple del agua, definido exactamente como 273,163 K o 0,01 ° C. Utiliza las mismas graduaciones que en la escala Celsius.
Tabla 4.1 Propiedades del agua
 09.17.40.png)
Los datos anteriores dan un ejemplo gráfico del comportamiento anómalo del agua; por ejemplo, se muestran temperaturas "anormales" de fusión y ebullición del agua. Sin embargo, esto está lejos de ser el único ejemplo de anomalía del agua. La razón de las propiedades anormales del agua son los enlaces de hidrógeno y las peculiaridades de la estructura del agua. El actual modelo de racimo del agua explica muchas de sus propiedades anormales. Por tanto, un puente de hidrógeno (enlace) es un elemento de enlace, un enlace de covalencia normal, llamado, entre una molécula y un enlace normal para la molécula de la fuerza de atracción débil de Van der Waals. En una gota de agua, miríadas de partículas forman una red infinita de tetraedros, mientras que los puentes de hidrógeno se organizan en un sistema ordenado. Al hacerlo, son flexibles y compatibles, lo que garantiza la variación del agua.
La extrema ligereza del hielo está relacionada con el hecho de que los puentes de hidrógeno tejen moléculas de agua en un cristal en redes volumétricas de tal manera que queda mucho espacio entre las partículas. Durante la fase de fusión, la red regular se rompe parcialmente y se convierte en líquido, lo que aumenta la densidad del agua.
El fenómeno antes mencionado se puede explicar discutiendo los enlaces de hidrógeno (puentes). La diversidad de formas macroscópicas sólidas de H2O se refleja a nivel molecular. Las variantes de los cristales de H2O demuestran una complejidad y sofisticación que no se pueden comparar con ninguna otra molécula. Como ya se ha señalado, se conocen 15 tipos de cristales. Christopher Zalzman de la Universidad de Oxford anunció los dos últimos descubrimientos en marzo de 2006. En el artículo[65], se presenta la información resumida sobre las propiedades de los 15 tipos de agua. Thomas Lepting, del Instituto de Química Inorgánica y Teórica General de la Universidad de Innsbruck, ha estado investigando el hielo desde 1980. “Incluso bajo presión normal, hay dos formas de hielo”, afirma. Estos son los llamados hielo hexagonal y cúbico, Ih e Ic, respectivamente[66]. En la Tierra se encuentra casi exclusivamente hielo hexagonal. Los copos de nieve, el hielo que cubre lagos y estanques y los cubitos de hielo en un vaso con una bebida consisten en este hielo. Sus partículas forman una estructura hexagonal. La forma básica del otro tipo de hielo es un cubo. Estos cristales, que tienen forma cúbica, solo se pueden encontrar en grandes alturas de la atmósfera terrestre, donde reina un frío mortal. Sin embargo, ambas variantes de hielo están unidas por una propiedad: la tridimensionalidad. No es sencillo tener en cuenta los posibles tipos de variedades cristalinas de hielo. El agua también es capaz de formar formas vítreas y amorfas. Los científicos creen que el 99,9% del hielo en el espacio está en forma amorfa. Cubre partículas de polvo en el espacio interestelar y se incorpora por composición de cometas.
La diferencia entre el hielo cristalino y el amorfo radica en sus estructuras internas. En un cristal, las moléculas de agua están dispuestas de manera regular en todas las direcciones con espacios intermedios idénticos. En el estado amorfo son caóticos, al igual que en el estado líquido, como si el líquido se hubiera solidificado instantáneamente. El representante más conocido de tal forma de materia se puede encontrar en un cristal de ventana ordinario. Según una interpretación científica, el hielo se ha convertido en la base de la vida en la Tierra. Su estructura flexible permite liberar elementos como nitrógeno, oxígeno y carbono. Al interactuar entre sí, estas sustancias forman biomoléculas simples. En la Tierra, se puede obtener hielo vitrificado universal si más de una centésima parte de una fracción de segundo es capaz de disminuir la temperatura en cientos de grados Celsius o en una prensa de alta presión. Un trozo de hielo obtenido en el Laboratorio de Innsbruck a una presión de 9,0 GPa con un enfriamiento brusco es de hecho hielo amorfo: HAD (hielo amorfo de alta densidad[67]). Externamente, no se diferencia en absoluto del hielo ordinario, pero si se echa en un vaso de agua, se hundirá porque es más pesado que el agua. Cuando se calienta, el HAD se transforma en una variante más de hielo vitrificado que, debido a su baja densidad, se denominó LAD (hielo amorfo de baja densidad[68]). Sin embargo, cuando los científicos calentaron HAD en la prensa, habiendo aumentado la temperatura de 196 a + 105°C, la muestra se contrajo. Esto contradice la idea generalmente aceptada de que las sustancias sometidas a una temperatura elevada deben expandirse. Por lo tanto, los científicos afirmaron que habían descubierto la tercera variante de hielo vitrificado: hielo amorfo de muy alta densidad (VHAD[69]).
El estado del agua cuando no hay diferencia entre líquido y gas se logra en el punto con una temperatura de 374 °C y una presión de 22.1 MPa. Los científicos creen que a bajas temperaturas también debería existir ese punto. En condiciones experimentales, se logra bajar la temperatura del agua a -38 °C, y en este caso no se congela. Tal fenómeno parece irreal, sin embargo, existe. Las nubes ligeras a una altitud de varios kilómetros sobre la superficie de la Tierra forman gotas y el líquido permanece aproximadamente a la misma temperatura (-38 °C). Cuando la temperatura disminuye, el agua "sobreenfriada" se comporta de una manera más extraña. Los científicos insinúan que a una temperatura de -50 a + 100 °C y una presión de 200 MPa se debería encontrar el segundo punto crítico[70]. Por debajo de esta marca, de acuerdo con la teoría, hay dos variedades de agua líquida con diferentes densidades; a mayor densidad, estas variantes no se distinguen.
Se supone que dos formas de hielo, HAD y LAD, corresponden a estas formas líquidas después de sus procesos de congelación rápida. Sin embargo, el resultado es que dentro de un rango de temperatura en el que, a pesar de todos los trucos de los experimentadores, las gotas de agua siempre se congelan. ¿Deberían existir varias formas de agua líquida? Esta es una de las muchas preguntas para las que todavía no hay una respuesta concluyente.
4.14 Anomalías del agua
Cabe señalar que las propiedades anómalas del agua, como las propiedades anómalas de las moléculas de oxígeno, son factores importantes para la existencia de la biosfera de la Tierra[71]. Entonces, ¿cuántas anomalías hay en el agua? Algunos científicos han contado 40 anomalías características del agua. Estos científicos han tratado de explicar estas propiedades anómalas del agua; a algunos les parecen agotadoras, a otros controvertidas y al resto insatisfactorias.
La importancia del papel de las propiedades anómalas del agua en la vida puede entenderse, por ejemplo, considerando sus propiedades especiales a 4 °C (Fig. 4.6). Para la determinación directa de la estructura del hielo y el agua líquida, se utilizan los datos del análisis de difracción de rayos X y la dispersión de neutrones lentos[72]. En estos métodos, la principal característica estructural es la función de distribución de la distancia entre los átomos de oxígeno. Para agua a 1.5 °C, el máximo de esta función corresponde a la distancia entre las moléculas de agua más cercanas en 0.290 nm, que supera ligeramente la distancia entre las moléculas del agua en el hielo (0.276 nm).
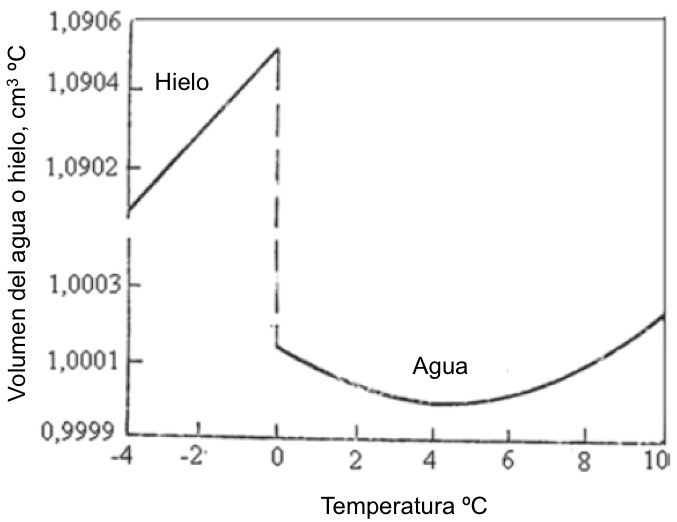
Fig. 4.6 Relación entre el volumen específico de hielo, agua y temperatura.
En este caso, el número de coordinación medio (n) de cada molécula de agua es igual a 4.4. En otras palabras, la destrucción de los enlaces de hidrógeno al derretir el hielo no implica un aumento de la densidad del agua líquida, y la razón de esto radica en un gran número de moléculas de coordinación. Esto se debe al hecho de que ocupan parcialmente huecos, que están vacíos en el hielo. Valores cercanos de las distancias entre moléculas de agua líquida y hielo ya en 1933 hicieron posible que varios autores expresaran la opinión de que el agua es un pseudocristal[73] (Fig. 4.5). A su vez, un autor logró explicar cualitativamente por qué la densidad del agua a 4 °C alcanza un máximo. Por tanto, de acuerdo con este modelo, el agua en estado líquido cercano a 0 °C representa una mezcla de tres componentes con diversas estructuras, que corresponden al hielo hexagonal, el cuarzo cristalino y la estructura densamente empaquetada del agua líquida. A medida que aumenta la temperatura, las estructuras parecidas al hielo se destruyen y aumenta la contribución de la estructura densamente compacta del agua. A ?> 4 °C, comienza a prevalecer el efecto de aumentar la distancia entre las moléculas. Está determinada por el calor causado por el movimiento que conduce a la disminución de la densidad del agua. En los años siguientes, se desarrollaron varios tipos de modelos de clústeres. Estos modelos permitieron explicar la mayor parte de las anomalías tanto del agua pura como del agua que contiene iones o moléculas[74].
La densidad máxima del agua a 4 °C, junto con una baja densidad del hielo, hacen que antes de la congelación sea necesario que todo el volumen de agua dulce (no solo su superficie) esté cerca de los 4 °C. Dado que la congelación de ríos, lagos y océanos se mueve de arriba a abajo, existe la posibilidad de que el ecosistema del fondo sobreviva, aislando el agua para que no se congele más, reflejando la luz solar en el espacio y descongelando la superficie del agua.
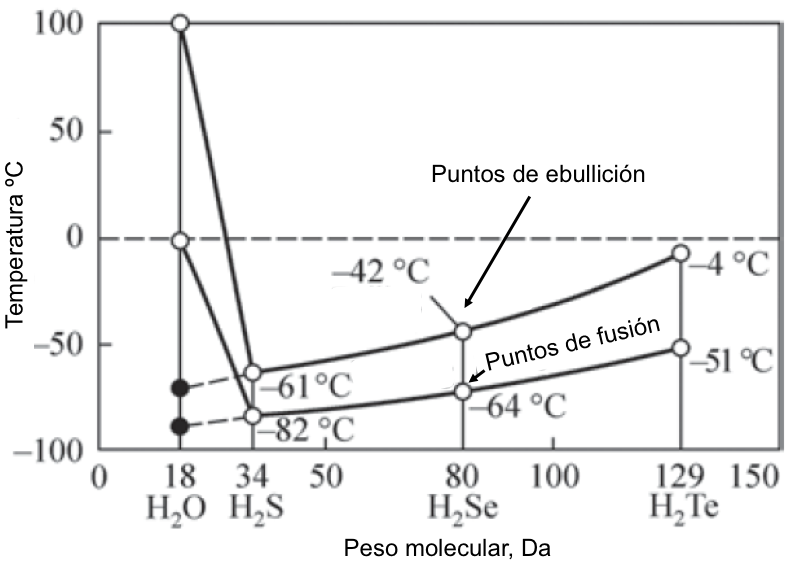
Fig. 4.7 Anomalías de los puntos de agua de ebullición y congelación en comparación con otros compuestos de hidrógeno
Gracias a la convección de calor, que está controlada por la densidad, se produce una mezcla estacional en aguas más profundas del clima moderado, lo que lleva el oxígeno que sustenta la vida a sus profundidades. La alta capacidad calorífica de los océanos y los mares hace posible que actúen como depósitos de calor de tal manera que las temperaturas marinas varían solo en un tercio. Esto es causado por el cambio de temperatura de la tierra, que determina nuestro clima (por ejemplo, la Corriente del Golfo lleva el calor tropical al noroeste de Europa). La compresibilidad del agua disminuye el nivel del mar en aproximadamente 40 m, lo que nos da un 5% más de tierra. Una alta tensión superficial del agua, más su expansión al congelarse, estimula la erosión de las piedras, formando así suelo para la agricultura.
Las propiedades opuestas del agua fría y caliente (ver Fig. 4.7) se señalan entre sus anomalías, que ya se han mencionado. Estas propiedades se expresan más vívidamente a temperaturas más bajas, cuando las propiedades del agua sobreenfriada se desvían del hielo hexagonal. Si el hidruro de oxígeno del agua ?2? fuera un compuesto monomolecular normal, como sus analogías del sexto grupo de DI Mendeleyev Periodic System of Elements, hidruro de azufre ?2S, hidruro de selenio ?2Se, hidruro de telurio ?2??, entonces el agua en estado líquido variaría de -90 a -70 °C. Con tales propiedades del agua, la vida en la Tierra no existiría.
A medida que aumenta la presión, las moléculas de agua fría se mueven más rápido, mientras que las moléculas de agua caliente se mueven más lentamente. El agua caliente se congela más rápido que el agua fría y el hielo se descongela en compresión, excepto bajo altas presiones, cuando el agua líquida se congela bajo compresión. No existe ningún otro material así en condiciones normales en forma de sólido, líquido y gas. Para toda la biosfera, una característica importante del agua es su capacidad, cuando se congela, no para disminuir sino para aumentar su volumen, es decir, para reducir su densidad. Esta anomalía del agua se conoce como anomalía de densidad. Inicialmente, esta fuente de agua atrajo la atención de G. Galileo. En la transición de cualquier líquido (excepto el galio y el bismuto) a un estado sólido, sus moléculas están dispuestas de manera más densa y la sustancia misma, mientras disminuye su volumen, se vuelve más densa. Esto es cierto para todos los líquidos excepto el agua.
Durante el proceso de enfriamiento, el agua se comporta al principio como otros líquidos; gradualmente se vuelve compacto y reduce su volumen. Este fenómeno se puede observar a + 4 °C (o para ser precisos, a + 3.984 °C) (ver Fig. 4.6). Es a una temperatura de + 3.984 °C que el agua tiene la mayor densidad y el menor volumen. El enfriamiento adicional del agua da como resultado gradualmente un aumento del volumen en lugar de una disminución.
La tendencia gradual de este proceso se termina repentinamente, y a 0 °C el volumen aumenta bruscamente en casi un 10%. En este instante el agua se convierte en hielo. Las características únicas del comportamiento del agua en los procesos de enfriamiento y formación de hielo juegan un papel excepcionalmente importante en la naturaleza y la vida. Estas características del agua evitan la congelación de todos los cuerpos de agua de la Tierra en invierno y, por lo tanto, salvan la vida acuática en la Tierra.
A diferencia del agua dulce, el agua de mar se comporta de manera diferente durante el proceso de enfriamiento. No se congela a 0 °C sino a -1.8 a -2.1 ° C, dependiendo de la concentración de sales disueltas en él. Tiene una densidad máxima de - 3.5 °C en lugar de + 4 °C. Así, se convierte en hielo sin alcanzar la mayor densidad. Si la agitación vertical en cuerpos de agua dulce termina enfriando toda la masa de agua a + 4 °C, entonces se produce la circulación vertical en el agua de mar a temperaturas por debajo de 0 °C. El proceso de intercambio entre las capas superior e inferior de agua procede como tal, creando continuamente condiciones favorables para el desarrollo de organismos animales y vegetales.
El agua de deshielo, formada por el derretimiento de glaciares e icebergs, es un medio especialmente favorable para los habitantes de los mares y océanos. Según sus propiedades termodinámicas, el agua se diferencia notablemente de otras sustancias. La más significativa entre estas sustancias es la anomalía del calor específico. La alta temperatura del agua convierte a los mares y océanos en un gigantesco regulador de nuestro planeta y, como resultado, se produce una marcada diferencia climática de invierno a verano y del día a la noche. El clima en las zonas costeras es templado; en estas áreas, las diferencias de temperatura a lo largo de las estaciones son bajas. Las poderosas corrientes atmosféricas que contienen enormes cantidades de calor se absorben en el curso de la vaporización, y las gigantescas corrientes oceánicas juegan un papel esencial en la creación del clima en nuestro planeta.
La anomalía del calor del agua se aclara en la siguiente descripción (Fig. 4.8). En el calentamiento de cualquier sustancia, la capacidad calorífica aumenta continuamente. Sin embargo, esto no es cierto para el agua. A medida que aumenta la temperatura del agua, su capacidad calorífica varía de forma anormal: de 0 a 37 °C, la capacidad calorífica disminuye y de 37 a 100 °C crece de manera constante. Para rangos de temperatura cercanos a 37 °C, la capacidad calorífica del agua es mínima.
Esta temperatura está cerca de la temperatura del cuerpo humano. La física del agua dentro del intervalo de temperatura de 35–41 °C (los límites de los procesos fisiológicos que ocurren normalmente y que son posibles en los organismos humanos) registra la probabilidad de lograr un estado único del agua; cuando las masas de agua cuasicristalina y volumétrica son iguales entre sí y la capacidad de una estructura para penetrar en la otra, se maximiza la variabilidad. Esta extraordinaria capacidad del agua predetermina la equiprobabilidad de ejecutar reacciones bioquímicas reversibles e irreversibles en los organismos humanos y asegurar un "fácil control" de las mismas.
Fig. 4.8 Anomalía del calor del agua
La excepcional capacidad del agua para disolver cualquier materia es de conocimiento común. Aquí demuestra anomalías extraordinarias para un líquido, a saber, las de la constante dieléctrica del agua. Esto está relacionado con el hecho de que la constante dieléctrica (o permitividad) constituye 81, mientras que para otros líquidos no excede de 10. De acuerdo con la ley de Coulomb, la fuerza interactiva de dos partículas cargadas en el agua será 81 menor que, por ejemplo, en el aire, donde esta característica es igual a la unidad.
En este caso, la fuerza de los enlaces intramoleculares se reduce en 81 veces y, bajo el efecto del calor, el movimiento se disocia con la formación de iones. Cabe señalar que, debido a su excepcional capacidad para disolver otras materias, el agua nunca es estrictamente pura.
Una anomalía maravillosa del agua es su tensión superficial increíblemente alta. De todos los líquidos conocidos, solo el mercurio tiene una tensión superficial más alta que el agua. Esta propiedad se manifiesta de hecho que el agua siempre intenta reducir su propia superficie. Los enlaces intermoleculares no compensados ??de la capa externa (superficial) de moléculas de agua, causados ??por fuerzas mecánicas cuánticas, crean la película elástica externa, que evita la inmersión de muchos objetos más pesados. Por ejemplo, si una aguja de acero se coloca con cuidado en el agua, no se hunde. Además, la densidad del acero es hasta ocho veces mayor que la densidad del agua. Debido a la alta tensión superficial, una gota de agua, por ejemplo, adopta una forma de bola en caída libre.
La tensión superficial y la humectación forman la base de una propiedad especial del agua y las soluciones acuosas, denominada capilaridad. La capilaridad es de gran importancia en la vida del mundo vegetal y animal, la formación de la estructura de los minerales naturales y la fertilidad de la tierra. En los canales muchas veces más estrechos que un cabello humano, el agua adquiere propiedades maravillosas: se vuelve más viscosa; se compacta 1.5 veces; se congela entre - 80 y - 70 °C. La causa del agua capilar sobre normal es la interacción intermolecular, cuyos secretos aún no se han revelado.
Los científicos y los expertos conocen la denominada agua porosa. Una película muy delgada recubre la superficie de los poros, microhuecos y minerales de la corteza terrestre y otros objetos de la naturaleza viva e inanimada. Unido a las fuerzas intermoleculares ya la superficie de otros cuerpos, posee una estructura especial, al igual que el agua capilar.
Según el agua se puede caracterizar por 63 anomalías[75]. Sea agrupado las propiedades anómalas del agua en los siguientes cinco grupos: anomalías de fase, anomalías de densidad, anomalías del agua como sustancia, anomalías termodinámicas y anomalías físicas. Anomalías de fase el agua tiene un punto de fusión, un punto de ebullición y un punto crítico inusualmente altos. En forma sólida, existe en una variedad más amplia de estructuras cristalinas y amorfas estables (y metaestables) que otros materiales. La conductividad del calor del hielo disminuye a medida que aumenta la presión, mientras que la estructura del agua líquida a alta presión varía. El agua sobreenfriada tiene dos fases y el segundo punto crítico está aproximadamente a -91 °C. El agua líquida se sobreenfría fácilmente pero se solidifica con dificultad; existe a temperaturas muy bajas y se congela al calentarla. El agua caliente puede congelarse más rápido que el agua fría (el efecto Mpemba) y el agua tibia vibra (fluctúa) más tiempo que el agua fría.
4.14.1 Anomalía de la densidad
La densidad del hielo aumenta cuando se calienta (hasta 70 K). El agua se comprime cuando se descongela (se derrite). La presión disminuye el punto de fusión. El agua líquida tiene una alta densidad, que aumenta cuando se calienta (hasta 3.984 °C). La presión disminuye la temperatura de la densidad máxima. El agua sobreenfriada tiene una densidad mínima. El agua se caracteriza por un bajo coeficiente de expansión (coeficiente térmico de expansión volumétrica — TCVE), que disminuye sustancialmente (se vuelve negativo) a bajas temperaturas. El TCVE del agua aumenta a medida que aumenta la presión. El número de vecinos más cercanos en hielo y deshielo (derretimiento) aumenta con el aumento de las temperaturas. El agua tiene una compresibilidad inusualmente baja, que disminuye con un aumento de la temperatura a 46,5 °C. En la relación de compresibilidad de la temperatura hay un máximo. La velocidad del sonido en el agua aumenta con el aumento de la temperatura a 74 °C. El "sonido rápido" se detectó a altas frecuencias; no es uniforme a una presión más alta. La RMN de la relajación de la red de espín es muy corta a bajas temperaturas. El índice de refracción del agua tiene un valor máximo a temperaturas inferiores a 0 °C.
4.14.2 Anomalía del agua como sustancia
Ninguna solución de agua es ideal. Los líquidos D2O y T2O difieren mucho del H2O en términos de sus propiedades físicas; H2O y D2O difieren en sus comportamientos de fase. Las sustancias disueltas afectan propiedades tales como la densidad y la viscosidad de manera diferente. La solubilidad de los gases no polares en agua disminuye con un aumento de temperatura al mínimo y luego aumenta nuevamente. La permitividad del agua es alta y demuestra la temperatura máxima. La movilidad de protones e iones de hidroxonio son anormalmente altos en el campo eléctrico. La conductancia del agua aumenta hasta un máximo de aproximadamente 230 °C. Las constantes de acidez de los ácidos débiles tienen mínimos de temperatura. La difracción de rayos X demuestra una estructura inusualmente detallada. Bajo altas presiones, las moléculas de agua se alejan aún más unas de otras a medida que aumenta la presión.
4.14.3 Anomalías termodinámicas
La variación máxima de temperatura del calor de fusión del agua es de -17 °C. El calor específico del agua es el doble que el del hielo o el vapor. El calor específico del agua (??, ?V) es inusualmente alto. El parámetro ?? es mínimo a 36 °C y máximo a aproximadamente -45 °C; hay un mínimo con respecto a la presión. El calor específico ?V tiene un máximo. También existen altas temperaturas de evaporación y sublimación; esta es la entropía de la evaporación. La capacidad calorífica del agua es alta y aumenta al máximo, que es aproximadamente 130 °C (Fig. 2.9).
Fig. 4.9 Dependencias de la temperatura de las características físicas del agua: 1 densidad, 2 viscosidad, 3 cambio de viscosidad con una presión, 4 compresibilidad, 5 capacidad calorífica Cp, 6 velocidad del sonido, 7 expansión térmica.
4.14.4 Anomalías físicas
El agua tiene una viscosidad inusualmente alta, que aumenta mucho cuando la temperatura desciende y disminuye con un aumento de la presión a temperaturas inferiores a 33°C. Al disminuir la temperatura, la difusión se debilita apreciablemente; a bajas temperaturas, la autodifusión del agua aumenta con el aumento de la presión y la densidad. La difusión de calor aumenta hasta un máximo de aproximadamente 0.8 MPa. El agua tiene una concentración-tensión superficial inusualmente alta (efecto Johnson-Ray) y previene la coalescencia de pequeñas burbujas.
Así es como el autor comenta sobre la clasificación de las propiedades anómalas del agua que propuso: “Que las propiedades del agua se vean o no como anómalas depende de qué materiales se va a comparar el agua y de la interpretación de 'anómala'. Por ejemplo, bien podría argumentarse que el agua posee exactamente esas propiedades que se podrían deducir de su estructura. Se considera que las comparaciones entre agua, sodio líquido, argón y benceno indican que varias de las propiedades antes mencionadas no son anómalas. Sin embargo, estos materiales quizás no sean los más típicos de los líquidos. Mi lista da las propiedades inusuales generalmente entendidas para hacer que el agua líquida (y el hielo) se destaque de los líquidos (o sólidos) “típicos”.
Por lo tanto, es muy difícil obtener agua pura (por ejemplo, <5 ng.g−1). Tenga en cuenta que el hielo, por el contrario, es un disolvente muy pobre y se puede utilizar para purificar el agua (por ejemplo, degasificar) mediante la implementación de sucesivos ciclos de congelación-descongelación.
Algunos científicos atribuyen la naturaleza anómala del agua a baja temperatura a la presencia de un segundo punto crítico; plantean una hipótesis interesante, aunque algo improductiva (ya que la atribución mezcla causa con efecto). Las anomalías del agua no requieren esto como explicación. El rango de temperatura de agua "caliente" a "fría" varía en estos ejemplos; consulte las entradas individuales para obtener más detalles.
Las anomalías del agua se dividen en grupos, pero es evidente que algunas anomalías pueden incluirse en más de un tema y es posible que no haya un acuerdo universal para las agrupaciones.
4.15 El papel del agua en el escenario de desarrollo en la Tierra
Una de las cuestiones más difíciles para las que no hay una respuesta inequívoca es la siguiente: ¿cómo apareció la vida en la Tierra? ¿Cuál podría ser un comienzo que hizo posible la vida en la Tierra? El presente estudio busca este inicio, al que se hace referencia en el artículo como “moléculas de protolife”, ya que el desarrollo de la ciencia cambia el escenario del surgimiento de la vida en la Tierra.
Entonces, ¿qué pasó hace 4.600 millones de años y cómo nació la vida? Para el científico genuino C. Darwin, las obras no son un dogma, sino una guía para la acción. La historia de la evolución contiene muchas ambigüedades, lo que no dice nada sobre la cuestión global del origen de la vida en la Tierra. En una de las últimas sesiones científicas de la Conferencia General SO RAN, el académico N. Dobretsov afirmó lo siguiente: la edad de la Tierra, como otros cuerpos del Sistema Solar, constituye aproximadamente 4.600 millones de años, lo cual ha sido confirmado por el análisis de meteoritos.
Dos conclusiones importantes que se pueden sacar hoy, basadas en numerosos datos, es el hecho de que la evolución de la vida está determinada por los procesos irreversibles de enfriamiento de la Tierra, que comenzó hace unos 4 mil millones de años, y la oxidación de las capas superiores, a saber, la hidroatmósfera, que se convirtió en el ímpetu para el comienzo de la evolución. Al descubrir la estructura del ácido desoxirribonucleico (ADN), los científicos comenzaron a buscar las moléculas responsables de la vida primitiva. El descubrimiento de la estructura del ADN se ha convertido en uno de los mayores logros científicos del siglo XX.
Fue realizado por un grupo internacional de investigadores: el P. Crick, J. Watson y M. Wilkins recibieron fotografías de rayos X de ADN de alta calidad; M. Nirenberg del Instituto Nacional de Salud Bethesda, Maryland, logró descifrar el código genético; R. Holy de la Universidad de Cornell aclaró la estructura del ARN de transferencia; un grupo de investigación del Instituto Tecnológico de Massachusetts, encabezado por el bioquímico indio G. G. Koran, sintetizó un gen artificial por primera vez en la historia del mundo. Todos los investigadores de estos experimentos recibieron premios Nobel. Por primera vez, la estructura del portador de información hereditaria, el ADN, se presentó en el artículo del genetista estadounidense J. Watson y del físico inglés F. Creek. De acuerdo con la definición de la American Chemical Society, el ADN son moléculas grandes que representan una doble hélice de átomos unidos y ubicados dentro de las células de todos los seres vivos capaces de crear, almacenar y transferir propiedades biológicas a generaciones sucesivas (el esquema de la estructura molecular del ADNse muestra en la figura 4.10).
Fig. 4.10 La estructura del ADN
El ácido desoxirribonucleico es un polímero cuyos monómeros son nucleótidos, una de las moléculas poliméricas más conocidas en la actualidad. En algunos organismos, la cadena de polímero de ADN consta de cien millones de enlaces. La longitud de dicha molécula alcanza varios centímetros, y este es un valor muy grande para los objetos moleculares. Dado que la sección transversal molecular es de solo 2 nm (1 nm = 10−9 m), sus proporciones se pueden comparar con un carril de decenas de kilómetros de largo.
La sucesión de polinucleótidos en una cadena de polinucleótidos sin ramificar, la estructura primaria del ADN, es estrictamente individual y específica para cada ADN natural. Representan una forma de código de registro de información biológica, conocida como código genético. En cada molécula, las cadenas de polinucleótidos están unidas en una doble hélice, en la que las bases están dispuestas en pares opuestos entre sí y unidas con enlaces de hidrógeno. Los pares se pueden formar de forma complementaria, es decir, con las bases emparejadas entre sí. La cadena se retuerce en una hélice alrededor de un eje imaginario con las bases perpendiculares a él. Esta doble hélice se llama estructura secundaria del ADN. La estructura terciaria está formada, por ejemplo, en los cromosomas, por dobles hélices unidas con proteínas por enlaces iónicos. La división celular provoca la duplicación o replicación del ADN (repleción, auto-duplicación) y, como resultado, cada nueva célula obtiene ADN que contiene la misma información genética, que estaba disponible en la célula inicial. La sucesión del ADN principal determina el orden de disposición de los residuos de aminoácidos en las moléculas de proteínas. Para la obtención de información genética basada en la cadena de codificación del ADN en el curso de la transcripción (la transferencia de la información genética del ADN al ARN), se forma una cadena de ARN, una réplica de la sección dada de ADN. La secuencia de bases de ARN es la matriz para la síntesis de proteínas codificadas en el propio gen.
Las proteínas fueron las primeras candidatas para el papel de “moléculas de protolife”. En 1924, A.I. Oparin afirmó que en la atmósfera de una Tierra joven que consta de hidrógeno, metano, amoníaco, dióxido de carbono y vapores de agua, los aminoácidos podrían sintetizarse y luego unirse espontáneamente en proteínas y formar "coágulos" que se asemejan a una célula primitiva.
A mediados de la década de 1960, los científicos conocían los detalles de dos moléculas en funcionamiento, que eran más adecuadas que las proteínas para el papel de "las moléculas de la vida protuberante", el ADN y el ARN[76]. La figura 4.11 muestra las estructuras de ADN y ARN para comparar. Las diferencias entre las macromoléculas de ARN y ADN se reducen al hecho de que el esqueleto de azúcar fosfato de ARN incluye azúcar ribosa, mientras que el ADN "pierde" un átomo de oxígeno y se transforma en desoxirribosa. Además, en lugar de timina (T), la composición del ARN incluye uracilo (U), que se diferencia de la timina casi tan poco como la ribosa y la desoxirribosa: carece únicamente del grupo metilo lateral (–CH3). Sin embargo, estas diferencias mínimas en las estructuras de ARN y ADN dan como resultado una diferencia sustancial en la estructura y funciones de estas moléculas.
A finales del siglo XX, se produjo un avance más en la teoría del origen de la vida. En este avance, se debe "culpar" al ARN, que se pensó que era una sustancia bastante investigada y predecible. Esta historia comenzó en la década de 1970, cuando se encontraron enzimas inusuales en las células de ciertos organismos. Las enzimas incluyen en su composición, además de la proteína, una molécula de ARN. A fines de la década de 1970, los bioquímicos estadounidenses Thomas Check y Sidney Altman, independientemente entre sí, estudiaron la estructura y funciones de tales enzimas. Una de sus tareas fue aclarar el papel del ARN incorporado como parte de las enzimas. Al principio, siguiendo la opinión recibida, los científicos creyeron que la molécula de ARN en tales complejos era solo un elemento auxiliar que puede ser responsable de la construcción de la estructura correcta de la enzima o de la orientación correcta en la interacción de la enzima y la enzima. El sustrato (es decir, esa molécula, que de hecho está sujeta al cambio) mientras que la reacción que se cataliza la realiza la proteína. Para aclarar la situación, los investigadores separaron los componentes de proteína y ARN entre sí e investigaron su capacidad para realizar catálisis. Para su gran asombro, vieron que incluso después de la eliminación de la enzima de la proteína, el ARN restante era capaz de catalizar su reacción específica. Tal descubrimiento significaría un gran avance en biología molecular. Anteriormente se creía que las reacciones solo podían ser catalizadas por proteínas en lugar de ácidos nucleicos.
Fig. 4.11 Estructura del ADN y el ARN
Sin embargo, ¿podría ser que se tratara de una purificación ineficaz del ARN, dejando algunos fragmentos mínimos intangibles de proteína que son capaces de implementar la catálisis? La última y más convincente prueba de la capacidad del ARN para la catálisis fue una demostración del hecho de que incluso el ARN sintetizado artificialmente, como parte de las enzimas en estudio, puede catalizar la reacción por sí solo. Las moléculas de ARN, capaces de catálisis, se denominaron ribozimas (por analogía con las enzimas, es decir, fermentos de proteínas).
Por su descubrimiento en 1989, T. Check y S. Altman recibieron el premio Nobel de química. Estos resultados no tardaron en arrojar luz sobre la teoría del origen de la vida; la molécula de ARN se convirtió en "la favorita". De hecho, ¡se había detectado una molécula capaz de transportar información genética y otro material para catalizar reacciones químicas! Sería difícil imaginar un candidato más adecuado para la generación de vida protocelular. Por tanto, el escenario de la evolución de la vida se ha transformado[77].
El escenario del comienzo de la vida en la Tierra consta de cuatro etapas, y al principio es el ARN (Fig. 4.12[78]). Dobretsov considera en detalle las cuatro etapas de la formación y evolución de la vida en la Tierra. ¿Tiene cabida en este escenario el agua como partícipe del origen de la vida en la Tierra? Con respecto a esto, la primera etapa como el comienzo mismo del origen de la vida en la Tierra es muy interesante.
En todos los sistemas vivos modernos, desde los virus hasta los animales superiores, el ADN o el ARN “disfrutan de los servicios” de las enzimas proteicas para transferir de manera rápida y eficaz, mediante catálisis, su información a varias generaciones. Ninguno de los ácidos nucleicos de los sistemas vivos modernos es capaz de replicarse.
La idea de un mundo compuesto de ARN, es decir, la existencia de ARN independiente en la primera etapa, que comenzó hace ~ 3.800 mil millones de años, se estableció hace menos de 20 años. En 1993, el Instituto de Proteínas de la Academia de Ciencias de Rusia (RAS) demostró empíricamente que las moléculas de ARN son capaces de formar colonias moleculares similares a las bacterias en geles u otros medios sólidos si se les proporcionan las condiciones para la replicación (repetición). Tales colonias moleculares sobre superficies sólidas o semisólidas (por ejemplo, montmorillonita con una película de agua en la superficie) consisten en conjuntos de moléculas de ARN con diferente actividad ribosómica y pueden ser los primeros conjuntos acelulares en evolución. La evolución de los conjuntos de ARN en colonias acelulares se aceleró debido a que estas colonias no estaban aisladas del medio ambiente y podían intercambiar fácilmente moléculas y su material genético.
Fig. 4.12 Las principales etapas de ocurrencia y evolución de la vida en la Tierra.
Se demostró la posibilidad de intercambiar las moléculas de ARN a través del aire debido a las recombinaciones espontáneas no enzimáticas de las moléculas de ARN en el caso de colisiones en el medio acuoso. Habiendo resumido las funciones conocidas, que son realizadas por el ARN en una célula, el autor del artículo de Dobretsov llegó a la conclusión de que todos los procesos básicos en la célula viva pueden implementarse solo mediante la presencia de diferentes tipos de ARN. Además, la famosa tríada ADN ↔ ARN → proteína tomó forma solo en la etapa I de la etapa II del origen y evolución de la vida en la Tierra, probablemente hace 3.600 millones de años (Fig. 4.12). La definición clásica de que “la vida es una forma de proteína celular de existencia de materia” significa que tanto las macromoléculas como la célula, es decir, la membrana con sus funciones complejas y variadas, son importantes para su existencia. Después de la aparición de la célula, el mundo bacteriano se desarrolló muy rápido.
Por tanto, ¿en qué fase del desarrollo de la Tierra, como planeta, se producen estas transiciones? Según la opinión de algunos científicos, el origen de la vida podría estar en el espacio. Esta teoría bastante popular de la panspermia afirma que la vida se transporta en todas las direcciones del Universo en forma de "esporas". Estos pueden ser compuestos orgánicos a veces de estructura bastante compleja, cuyas huellas se encontraron en los meteoritos que habían caído sobre la Tierra. Según los astrofísicos, la materia orgánica existe en grandes cantidades en las nubes de gas y polvo y en el polvo espacial, que constantemente es engullido por la Tierra. Según el autor del artículo de Dobretsov, la vida desde el espacio se transporta únicamente en forma de oligonucleótidos cortos, que pueden congelarse en hielo de cualquier composición (metano, acuoso) y transformarse en un medio favorable. Este ciclo comienza de nuevo cada vez: la síntesis de la montmorillonita, la aparición de las moléculas de ARN y ADN y la proteína, y finalmente, la emergencia de una célula.
Los científicos tienden a llegar a la conclusión de que la presencia de agua en los objetos espaciales es probablemente una regla, más que una excepción. Las sondas espaciales estadounidenses detectaron agua en dos de los satélites de Saturno y en Marte. A partir de esta evidencia, se puede asumir la existencia de vida en estos planetas en el pasado o en el presente. En 2001, los científicos del IMS Scientific and Research Center de la NASA y la división de la Universidad de California en Santa Cruz (EE. UU.) Llevaron a cabo un experimento en condiciones similares a las de la formación del Sistema Solar. La mezcla de diversas sustancias (agua, metanol, ácido carbónico y dióxido de carbono) se enfrió a 10 ? (-263.16 °C). Fue irradiado por luz ultravioleta con una longitud de onda similar a la longitud de onda en una densa nube molecular a partir de la cual supuestamente se formó el Sistema Solar. Se produjo la formación de moléculas orgánicas. Se identificaron estructuras autoorganizadas de 10 μm de tamaño. Por su forma, eran burbujas, que se parecían a células. En julio de 2005, los científicos estadounidenses encontraron al menos tres áreas cubiertas de hielo en la superficie del núcleo del cometa Temple. Según los investigadores, el agua, que se convirtió en hielo, contiene muchas impurezas. Los científicos llegaron hace mucho tiempo a la conclusión de que los cometas son fragmentos del Sistema Solar inicial en un instante de su inicio hace 4.600 millones de años.
Una conclusión importante, que se puede llegar hoy a partir de numerosos datos, es el hecho de que la evolución de la vida está determinada por los procesos irreversibles del enfriamiento de la Tierra, que comenzó hace unos 4 mil millones de años, y por la oxidación de las capas superiores, en primer lugar, la hidrosfera, que sirvió como causa principal del inicio de la evolución. Los científicos de un laboratorio dependiente del Departamento de Energía de los Estados Unidos llegaron a una conclusión sensacional en el curso de una investigación relacionada con un intento de explicar el cambio de cada 200 mil años en la dirección del campo magnético de la Tierra[79]. En el centro de la Tierra hay un núcleo de la mezcla fundida de uranio y plutonio, que mantiene una reacción nuclear constante, en lugar de hierro y níquel fundidos. Este núcleo, de casi 8 km de diámetro, es un “gigantesco reactor nuclear natural”. Como resultado de su “trabajo” alrededor de la Tierra, aparece un poderoso reactor nuclear, que protege al planeta de peligrosos rayos cósmicos capaces de aniquilar toda la vida biológica en unos segundos. El reactor natural también suministra energía al movimiento de las plataformas continentales y se manifiesta en la erupción de volcanes. Es obvio que se necesitan una serie de condiciones adicionales para el inicio del proceso. Uno de ellos es la presencia de agua. El agua (12-15%) es necesaria como moderador de neutrones, que se forman en el caso de la división espontánea del uranio, ya que la reacción puede ocurrir solo bajo el efecto de neutrones lentos.
Una hipótesis más interesante radica en el hecho de que la vida se originó cerca de las emisiones de agua volcánica caliente, donde, debido a la temperatura y la presencia de grandes concentraciones de moléculas biogénicas, la reacción de formación de biomoléculas puede ocurrir a altas velocidades de acuerdo con Dobretsov. Además, diferencias sustanciales de temperatura podrían facilitar los procesos de síntesis matricial de ácidos nucleicos. Las altas temperaturas (cerca de la fuente) condujeron a la descomposición de ácidos nucleicos de doble filamento en ácidos unifilares, durante los cuales (como resultado de una disminución de la temperatura) puede ocurrir el otro ciclo de síntesis. Tal escenario se asemeja a la tecnología de la replicación repetida de ácidos nucleicos, conocida como reacción en cadena de la polimerasa (PCR), desarrollada a mediados de la década de 1980[80].
Hasta ahora, la teoría del mundo del ARN está llena de contradicciones y ambigüedades. Muchas de ellas, sin duda, se resolverán en el marco de la hipótesis "clásica" que implica que la fuente de la vida era el ARN tal como lo conocemos en la forma moderna. Sin embargo, ¿cómo podemos explicar en el marco de esta teoría la evidencia de arqueólogos y paleontólogos que han encontrado los restos de las primeras células primitivas en capas del período de hace 3.5 a 3.8 mil millones de años? Al mismo tiempo, se puede considerar que la vida no pudo originarse antes de hace 4 mil millones de años porque antes de eso la Tierra había sido intensamente "bombardeada" por meteoritos y cometas. Según datos más radicales, el "bombardeo" terminó incluso más tarde, hace unos 3.800 mil millones de años. Por tanto, efectivamente no hubo tiempo para el desarrollo del mundo precelular. Los partidarios prominentes y fundadores de la hipótesis del mundo del ARN Thomas Chech y Lesley Orgel[81] estuvieron de acuerdo con esto.
Se han planteado dos hipótesis bastante atrevidas sobre el origen de la vida[82]. En primer lugar, los planetas, según se formaron en las condiciones del proceso de reacción catalítica. Se formaron a partir de una nube de gas y polvo, que parecía un enorme reactor catalítico, ya que en ausencia de reacciones químicas, las partículas de polvo no pueden mantenerse juntas. En segundo lugar, la selección natural comenzó ya en la etapa química de la evolución, mientras que los compuestos orgánicos primarios, de los cuales puede originarse la vida, ocurrieron antes de la formación de los planetas. Ninguna de las teorías existentes sobre el origen de la vida aclara cómo aparecieron los primeros ARN y ADN.
La probabilidad matemática de la "autocreación" aleatoria de estas moléculas es demasiado débil. De ahí que aparecieran numerosas especulaciones sobre el origen extraterrestre de la vida. Sin embargo, el inicio de la evolución podría desencadenarse por una reacción catalítica. El esquema más simple de tal reacción es el siguiente: la molécula de "alimento" más la molécula de un "asistente" (autocatalizador) es proporcionada por dos moléculas de autocatalizador.
En el curso de experimentar con la concentración de "alimento" y asumiendo que las moléculas del autocatalizador son capaces de mutar, uno puede encontrar un análogo de la selección natural; las moléculas que pueden hacerlo con una cantidad mínima de "alimento" pueden sobrevivir. Esto no se corresponde con la conocida teoría del “caldo espeso”, según la cual la vida se originó por el exceso de nutrientes. Aun así, la suposición de que el hambre es una "fuerza motriz del progreso" parece bastante lógica.
Sin embargo, ¿existe una reacción en la ciencia química que se corresponda exactamente con este ejemplo? La respuesta es sí, hay al menos uno. La reacción de Butlerov fue descrita por un destacado químico ruso ya en 1864[83]. Se trata de una síntesis de varios azúcares a partir del formaldehído que aparecen en presencia de iones de calcio o magnesio. Es bien sabido que la reacción de Butlerov es autocatalítica, aunque aún se desconoce la naturaleza y el mecanismo de acción de los autocatalizadores. La reacción es incontrolable; cada vez se obtienen azúcares muy diferentes en proporciones muy diferentes.
Se considera un hecho que la ribosa (un tipo de azúcar) en la reacción de Butlerov aparece de manera constante. Además, la ribosa es la base para la creación tanto de ARN como de ADN. De ahí que se desprenda una conclusión importante: las "piezas de repuesto" necesarias para el montaje del primer ARN y ADN podrían aparecer en el curso de la reacción de Butlerov. Además, como creen los geólogos, había una atmósfera explosiva compuesta de metano, vapor de agua, dióxido de carbono y amoníaco en la Tierra primitiva. Con tal cóctel para la formación de formaldehído, que es necesario en la reacción anterior, solo se necesitaba la presencia de una superficie caliente. Por lo tanto, la base de la vida no podría ser la proteína en absoluto, como es una creencia comúnmente aceptada; en cambio, probablemente fue el azúcar, que apareció en el curso de la reacción autocatalítica.
Basado en su propio modelo politetramérico de la estructura del agua, de la etapa más temprana en el desarrollo de la vida en la Tierra[84]: se dio la síntesis de materia orgánica prebiológica. Se considera que la participación del agua no solo como un solvente universal y único posible, sino también como una matriz que se forma cuando el agua participa en la codificación de proteínas a través de las bases nitrogenadas del ADN. Detrás de este enfoque, se encuentra el hecho de que se “logró revelar gráficamente la existencia de tetrámeros acuosos izquierdos y derechos jugando en el agua habitual en el papel de moléculas... donde la simetría bilateral de las unidades estructurales del agua ... debería afectar de alguna manera la síntesis prebiológica de la materia orgánica”. El escenario propuesto en el artículo[85] de la etapa inicial en el desarrollo de la vida en la Tierra se basa en el fenómeno de la aleatoriedad y en ausencia de datos experimentales sobre la existencia de simetría bilateral de las unidades estructurales del agua en condiciones reales, cuando la vida se originó en el planeta Tierra.
Según las ideas contemporáneas sobre el origen de la vida en la Tierra, la elección elegida por moléculas orgánicas de cierto tipo de simetría bilateral sirvió como prerrequisito principal para su supervivencia y posterior reproducción. Sin embargo, la cuestión de cómo y por qué ocurrió la selección evolutiva de esta u otra antípoda espejo sigue siendo uno de los mayores acertijos de la ciencia. El científico soviético L. L. Morozov demostró que una transición al orden quiral podría tener lugar en el caso de un cierto cambio de fase brusco en lugar de un cambio evolutivo[86]. El académico V. I. Goldansky se refirió al período durante el cual se originó la vida en la Tierra como una catástrofe quiral[87]. Entonces, ¿cómo aparecieron las condiciones para la fase catastrófica, que causó la transición quiral? Lo más importante fue el hecho de que los compuestos orgánicos se derritieron a 800-1.000 °C en la corteza inferior de la Tierra, mientras que los compuestos de la corteza superior se enfriaron a la temperatura del espacio, es decir, el cero absoluto.
En tales condiciones, la diferencia de temperatura alcanzó los 1.000 °C. Las moléculas orgánicas de la corteza inferior se derritieron bajo el efecto de las altas temperaturas y fueron completamente destruidas, mientras que la capa superior de la corteza, donde las moléculas orgánicas se congelaron, permaneció fría. Los gases y vapores de agua que se filtraron de la corteza terrestre variaron la composición química de los compuestos orgánicos. Los gases transportaron calor, lo que provocó que el límite de fusión de la capa orgánica se desplazara hacia arriba y hacia abajo, creando un gradiente.
A presiones atmosféricas muy bajas, el agua en la superficie de la Tierra estaba solo en forma de vapor y hielo. Cuando la presión alcanzó el llamado punto triple del agua (60 Pa), el agua por primera vez podría estar en forma líquida. Ciertamente, solo empíricamente se puede probar qué causó la transición quiral: razones terrestres o espaciales. Sin embargo, en cualquier caso, en algún instante las moléculas ordenadas quirales (a saber, los aminoácidos que giran a la izquierda y los azúcares que giran a la derecha) resultaron ser más resistentes, lo que conduce al crecimiento continuo de su cantidad, lo que se conoce como transición quiral[88].
Lo más maravilloso de la estructura del agua radica en el hecho de que sus moléculas a bajas temperaturas negativas y altas presiones dentro de los nanotubos pueden cristalizar en forma de una doble hélice que se asemeja al ADN. Esto fue probado por experimentos informáticos de científicos estadounidenses, encabezados por Syao Chen Zen en la Universidad Estatal de Nebraska. Los científicos esperaban ver que el agua en todos los casos forma una estructura tubular delgada. Sin embargo, el modelo demostró que, dado el diámetro del tubo de 1.35 nm y la presión de 4.0 GPa, los enlaces de hidrógeno se deforman, lo que da como resultado la formación de una hélice de doble pared. La pared interior de esta estructura es una hélice cuadruplicada; el exterior consta de cuatro dobles hélices como la estructura de la molécula de ADN[89]. El último hecho afecta no solo a la evolución de nuestros conceptos sobre el agua, sino también a la evolución de la vida temprana y a la propia molécula de ADN. Si se asume que en la época del origen de la vida las rocas de arcilla criolítica tenían forma de nano-tubos, entonces surge una pregunta: ¿podría el agua absorbida en ellas servir como base estructural (matriz) para la síntesis de ADN y lectura de la información? Posiblemente, por lo tanto, la estructura helicoidal del ADN repita la estructura helicoidal del agua en nanotubos. Confirmar o refutar la existencia de tales macromoléculas solo puede conducir a investigaciones experimentales similares en las condiciones ambientales.
4.16 Composición isotópica del agua: el papel del deuterio en la destrucción del ADN
Solo en 1931 se conoció la existencia de agua pesada en la naturaleza. Su composición, aunque en cantidades muy pequeñas, incorpora los isótopos pesados ??del hidrógeno: deuterio y tritio. Este año estuvo marcado por el descubrimiento de hidrógeno pesado, deuterio, por el físicoquímico estadounidense Harold Urey. H. Urey encomendó a su asistente que evaporara 6 L de hidrógeno líquido, y en la última fracción de 3 cm3 por análisis espectral, detectó un isótopo pesado de hidrógeno con una masa atómica dos veces mayor que la del protio conocido[90].
Los científicos llegaron a la conclusión de que quizás existía un isótopo pesado de hidrógeno con una masa atómica de 2. En 1932, H. Urey y E.F. Osborn encontraron por primera vez en agua natural uno pesado[91]. Dos años después, Harold Urey recibió el Premio Nobel. El descubrimiento del tercer isótopo de hidrógeno superpesado, el tritio, con masa atómica 3, se mantuvo en secreto durante 3 años debido a consideraciones estratégicas. En 1951, se obtuvo e investigó el agua con tritio. En agua natural, la fracción atómica de tritio es insignificante con solo 10-18[92].
No obstante, está contenido en el agua que usamos para beber y, con el paso de los años, daña sustancialmente nuestros genes, provocando envejecimiento y enfermedades. El agua con deuterio se obtiene con una minúscula cantidad de agua de tritio por su concentración en los restos de electrolito después de la descomposición electrolítica del agua natural y también en la destilación fraccionada de hidrógeno líquido. La fabricación industrial de agua pesada aumenta cada año en casi todos los países, y especialmente en los estados que poseen armas nucleares.
El agua pesada se utiliza principalmente como moderador de neutrones rápidos en la división de elementos radiactivos en reactores nucleares. Las perspectivas de utilizar agua pesada o necesidades humanas son grandiosas. Puede convertirse en una fuente inagotable de energía: 1 g de deuterio proporciona decenas de millones de veces más energía que la quema de 1 g de carbón.
El agua de tritio hasta ahora tiene un uso limitado y se utiliza, principalmente, en reacciones termonucleares y también en investigaciones fisicoquímicas y biológicas como moléculas radioactivas trazadoras. Se han detectado seis nucleidos de oxígeno: 14?, 15?, 16?, 17?, 18?, 19?. Tres de ellos — 16?, 17?, 18? son estables, mientras que 14?, 15? y 19? son radiactivos. Toda el agua natural contiene isótopos de oxígeno estables. Sus proporciones son las siguientes: por 10000 porciones de 16? caen 4 partes de 17? y 20 partes de 18?.
¿Cuál es el agua de la Tierra desde el punto de vista de la composición isotópica de los elementos que la forman? Para la caracterización de la composición del isótopo del agua, se utilizan tres estándares: el estándar generalmente aceptado SMOW, y también GISP y SLAP, que se dan a continuación.

El estándar SMOW (Standard Mean Ocean Water) corresponde a las aguas profundas del océano mundial con un contenido de 997,0325 g/kg (99,73%) de agua ligera 1H2 16O. El estándar SLAP (Precipitación Antártica Ligera Estándar) corresponde al agua natural de la Antártida, donde la fracción de agua ligera constituye 997,3179 g/kg (99,76%). La composición isotópica del agua en la Tierra, teniendo en cuenta solo los isótopos estables, es la siguiente:

Como puede verse en los datos mostrados, la combinación de isótopos estables produce nueve variedades de moléculas. En este caso, el agua a granel cae sobre las moléculas de agua con protio (ligera) con oxígeno del 16 al 99,727% y sobre las moléculas más pesadas solo el 0,273%.
En este caso, las moléculas pesadas se distribuyen de la siguiente manera: ?218?—73,3; ?217? — 14,7; HD16O es el 12,1% del volumen total. En las fuentes de agua dulce, el contenido de agua pesada suele constituir unos 330 mg/dm3 (por molécula de HDO) y en una molécula de oxígeno pesado (?218?), unos 2 g/dm3. Esto se puede comparar o incluso superar el contenido permitido de sales en el agua potable. Además, se observó que las variaciones isotópicas naturales de D y 18O son anormalmente altas. Si consideramos el deuterio como un microelemento, incorporado no solo al agua, sino también a importantes compuestos orgánicos, entonces, en términos de importancia, puede clasificarse como uno de los primeros, si no el primero. Entre los otros elementos del organismo humano, el deuterio aparece inmediatamente después del sodio. La cantidad de deuterio en el plasma sanguíneo es cuatro veces más alta que el potasio, 6 veces más alta que el calcio, 10 veces más alta que el magnesio y mucho más alta que el contenido de oligoelementos tan importantes como el flúor, el hierro, el yodo, el cobre, el manganeso y el cobalto.
 14.41.37.png)
Fig. 4.13 Composición isotópica del cuerpo humano y número de átomos de los elementos que lo forman (unidad: número de átomos × 1024)
Como se muestra basado en las variaciones naturales del deuterio, se puede inferir que su contenido en el organismo humano varía mucho, de 9 a 16 mmol/dm3, incluso si se considera que durante la vida no se acumula en los organismos, debido a reacciones de intercambio isotópico; sin embargo, quizás no sea así. En el plasma sanguíneo humano, la concentración de deuterio es más alta que en el agua de bebida consumida, lo que se confirma con los siguientes datos[93]:
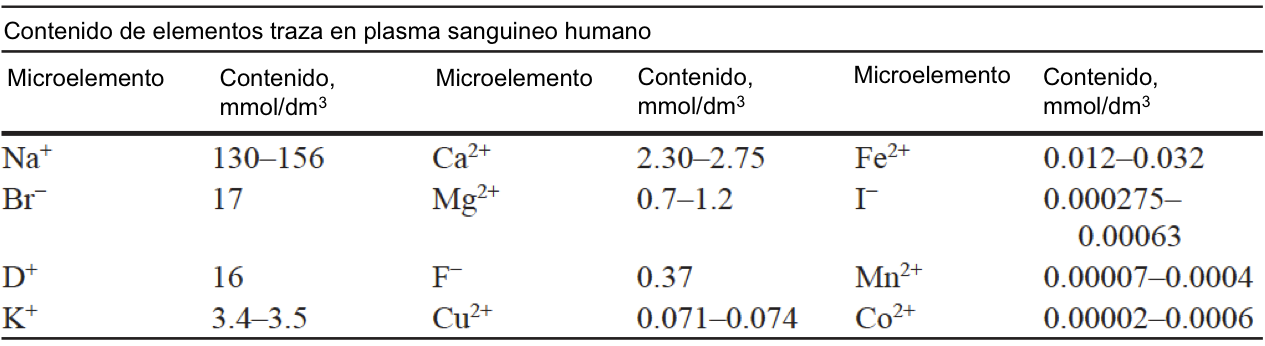
En plasma, el contenido de deuterio supera los 16 mmol/dm3. Dado que el hidrógeno no solo es parte del agua, sino también macromoléculas, que forman proteínas, grasas e hidrocarburos. Por lo tanto, se puede suponer que estas oscilaciones no serán ignoradas por el organismo, de modo que por el número de átomos en un cuerpo humano, el hidrógeno ocupa de manera convincente el primer lugar, aunque por la masa es solo el tercero (Fig. 4.13).
El agua pesada en la actualidad se ha estudiado suficientemente. Numerosas investigaciones que involucran isótopos pesados ??y radiactivos de oxígeno han demostrado que el agua, enriquecida con deuterio y tritio, es dañina para todas las formas de vida. Los efectos biológicos del agua pesada se manifiestan en: una reducción en la tasa de reacciones bioquímicas, la respiración de los tejidos, un aumento de la viscosidad del protoplasma de las células y la tasa de envejecimiento del organismo; otras reacciones incluyen la inducción de mutaciones, la lesión del acervo genético, enfermedades como el cáncer, la inhibición de la división celular, la reducción de la tasa de crecimiento y la muerte de vertebrados superiores.
El hecho de que el 97% de la masa de agua esté representada por isótopos ligeros y estables H y O supone una ventaja para la vida. El momento "mejor" del agua con tritio aún no ha llegado porque hay muy poco en la Tierra, solo unos 25-30 kg. Sin embargo, su cantidad en el agua de la Tierra crece continuamente ya que se forma durante el bombardeo de los núcleos de nitrógeno y oxígeno con rayos cósmicos. A diferencia del protio y el deuterio, el tritio es un elemento radiactivo con un período de vida media de 9 años. Las propiedades del agua de tritio superpesado difieren de las del agua de protio ligera más que del agua de deuterio.
El agua ligera es el agua empobrecida en deuterio cuyas moléculas contienen isótopos pesados ??de hidrógeno y oxígeno en cantidades mínimas. En la primera aproximación, dicha agua puede considerarse como una sustancia con la fórmula química H216O[94]. El agua de protio H216O en forma pura, es decir, sin las impurezas infinitesimales de las otras variedades de isótopos, debe considerarse clásica. Los isótopos de hidrógeno estables con isótopos de oxígeno estables forman 9 variedades de isótopos de moléculas de agua, a saber: H216O, H2 17O, H2 18O, HD16O, HD17O, que consta de nucleidos ligeros HD18O, D216O, D217O, D218O. En un aspecto cuantitativo, la mayor parte del agua de fuentes naturales está representada por las moléculas de H216O, que consisten en los nucleidos ligeros D, 17O y 18O. La cantidad de moléculas de agua que contienen nucleidos pesados ??D, 17O y 18O depende de la concentración de estos nucleidos, que en el agua natural varía dentro de los límites registrados en los principales patrones de composición isotópica de la hidrosfera. Aunque el contenido de agua de protio excede sustancialmente el contenido de todos los tipos tomados en conjunto, no se puede encontrar H216O puro en estas condiciones ambientales. El agua con una fracción sustancialmente aumentada de H216O, tiene un peso molecular menor, posee una densidad más baja y es la más ligera en la categoría de agua ligera isotópica. Tal agua se define como especialmente pura.
4.17 Características del agua ligera
La característica principal del agua ligera es la pureza isotópica de la sustancia, a diferencia del agua corriente. El agua ligera no contiene impurezas como agua pesada (D2?) y semipesada, cuyo peso molecular supera las 18 amu. El agua que contiene H216O presenta propiedades diferentes a las del agua con un contenido natural de isotopos.
Como medio universal, donde se dirigen todas las reacciones biológicas, el agua con un mayor contenido de H216O acelera estas reacciones, en comparación con el agua de composición isotópica natural. Este efecto se conoce en la ciencia fundamental como "el efecto isotópico cinético de un disolvente". Durante este efecto, los isótopos pesados ??inhiben o retrasan las reacciones más que los isótopos ligeros. Cuando los isótopos pesados ??se eliminan del agua, desencadena las reacciones; por lo tanto, el agua ligera generalmente activa reacciones biológicas de los sistemas vivos. Esta propiedad del agua ligera es útil en condiciones en las que la tasa de reacciones metabólicas es baja; se puede aplicar, por ejemplo, en el envejecimiento, enfermedades metabólicas como la diabetes, la resistencia a la insulina y el síndrome metabólico. La densidad y las temperaturas de las transiciones de fase del agua ligera es menor que la del agua pesada (Tabla 4.1).
Tabla 4.1 Cambio de las propiedades del agua bajo la sustitución de isótopos
 12.11.43.png)
La presión de equilibrio del vapor de agua de diferentes composiciones isotópicas difiere y atrae una atención aguda. Cuanto más ligera sea una molécula de agua, mayor será la presión del vapor, y esto significa que el vapor, que está en equilibrio con el agua, siempre está enriquecido con isótopos ligeros de oxígeno e hidrógeno. La reacción de los biosistemas cuando se someten al agua puede variar según los cambios cuantitativos y cualitativos del isótopo H2O. Efectos biológicos del agua ligera:
• Optimización de la velocidad de reacciones biológicas;
• Estimulación de la división celular y el crecimiento de organismos;
• Efecto radioprotector;
• Efectos antimutagénicos;
• Efecto médico-profiláctico y anticarcinomatoso.
4.17.1 Producción de agua ligera
Para la producción de agua ligera isotópica, los métodos y unidades propuestos tenían como objetivo reducir la cantidad de hidrógeno de isótopos pesados, principalmente deuterio, en el agua, preservando la composición isotópica inicial del oxígeno en H2O.
El método de obtención de agua de "fusión" y "relicto" con un contenido reducido de isótopos pesados ??de deuterio y tritio incluye las operaciones de enfriamiento de agua y posterior descongelación del agua congelada. Sin embargo, el grado en el que se puede reducir la cantidad de deuterio en este caso es bastante pequeño. Mediante el método de electrólisis se consigue un agotamiento bastante intenso del agua por los isótopos de deuterio, pero la capacidad de dicho aparato es muy baja.
Aunque la reducción del resto de deuterio en el agua da como resultado un aumento de la fracción de H216O, el agotamiento del deuterio en el agua solo limita la posibilidad de aumentar aún más el H216O. Un aumento sustancial del H216O en el agua es posible solo después de eliminar el número máximo de moléculas que contienen nucleidos pesados ??D, 17O y 18O. Por lo tanto, es preferible que el contenido de H216O en el agua producida comercialmente aumente debido a la eliminación de moléculas que contienen no solo deuterio sino también nucleidos de oxígeno pesados ??(17O, 18O).
4.18 El papel del deuterio en la destrucción del ADN
Los efectos isotópicos del agua pesada en altas concentraciones, que no existen en condiciones de campo, se estudian con éxito en la actualidad. La extrapolación de los datos obtenidos a una fuerte dilución de agua pesada no permite efectos tangibles en la reducción de la concentración de isótopos pesados ??por debajo del nivel natural. No obstante, en la última década se ha demostrado que el agua natural, empobrecida por isótopos pesados ??de hidrógeno y oxígeno, posee un efecto estimulante para diferentes objetos biológicos e incluso contiene propiedades medicinales[95]. De ese modo, se confirmó que una célula viva es capaz de responder a cambios infinitesimales del contenido de deuterio en el agua.
Ya en 1974, se expresó la suposición de que el deuterio era una posible razón del envejecimiento. Según los datos disponibles, el envejecimiento está relacionado con la acumulación gradual de errores de ADN que surgen en relación con la ruptura de la hebra, con los errores ocurridos durante la replicación del ADN o con la disfunción de los mecanismos de su reparación. Se consideró factores que afectan desfavorablemente al ADN y también representan los resultados de la investigación en la eliminación de estos factores del medio celular[96]. Se cree que el mutágeno más abundante, que daña el ADN, es la radiación solar de bajo nivel; el ADN también se ve afectado negativamente por el agua pesada (D2?, óxido de deuterio). La concentración de agua pesada en la superficie de la Tierra constituye 155 moléculas por millón. Debido a esta baja concentración, el agua pesada no suele llamar la atención. Sin embargo, las últimas investigaciones han demostrado que el óxido de deuterio, que generalmente se ignora, puede desempeñar un papel clave en el proceso de envejecimiento. Uno de los principales factores que limitan la vida humana es el daño natural del ADN, tanto por el efecto directo de la radiación como por los radicales libres producidos por la radiación, mutágenos y actividades metabólicas naturales.
La atmósfera de la Tierra (ozono) protege la mayor parte de la radiación cósmica; sin embargo, cierta cantidad de radiación aún penetra en la atmósfera y llega a la superficie de la Tierra. Esta radiación, junto con las fuentes radiactivas naturales de la Tierra, crea una radiación ionizante que afecta negativamente al ADN. En un cuerpo humano, casi todo el daño del ADN se elimina gracias a un mecanismo de reparación inherente (autorrecuperación). No obstante, en algunos casos, dichos mecanismos se alteran, lo que provoca enfermedades y estimula el envejecimiento.
Sin duda, enfermedades como el cáncer y una disminución total de la integridad del ADN están interrelacionadas. Esto significa que al final del ciclo de vida un ser humano "encuentra una barrera" cuando la integridad del ADN es controvertida y no existe un método para restaurar todas sus hebras, lo que conduciría a prolongar la longevidad. En consecuencia, existe una gran ventaja en la preservación del ADN desde una edad temprana para optimizar tanto la longevidad como la calidad de vida.
En artículos que tratan sobre el impacto biológico de la radiación de bajo nivel, se ha demostrado que el cambio gradual del ADN causado por la radiación y otros mutágenos es imposible de detectar con métodos contemporáneos. Se encontró que un efecto de radiación intensivo de corta duración da como resultado un envejecimiento prematuro, pero aún continúa la discusión sobre si las mutaciones son causadas por niveles normales de radiación. La radiación ionizante da como resultado la formación de radicales libres, que atacan al ADN, pero esto también ocurre durante los procesos metabólicos naturales. Algunos científicos incluso afirman que la radiación de bajo nivel desencadena una "respuesta inmune", que protege el ADN de daños adicionales. Los niveles naturales de radiación ionizante producen un efecto negativo débil, pero una cantidad muy pequeña de roturas de hebras de ADN restauradas incorrectamente, acumuladas durante décadas, puede causar efectos de envejecimiento[97].
Deuterio, mitosis y ADN. La división celular se encuentra en el centro del crecimiento y la propagación asexuada de los organismos. Es bien sabido que el alto nivel de deuterio retrasa la tasa de mitosis (división celular directa), pero aún no está claro un mecanismo preciso de este proceso. En 1989, Jan Lamprecht, Diter Schreter y Nidhart Pavelets llevaron a cabo una investigación sobre el impacto del deuterio en la mitosis en el Instituto de Biología Celular y Tumoral y en el Centro Alemán de Investigación del Cáncer en Heidelberg. En uno de los experimentos, las células fueron sometidas al tratamiento de agua pesada (óxido de deuterio) con concentraciones de 25, 50 y 75% durante 2 h, mientras que en el otro, por el impacto de un 75% de óxido de deuterio durante 2 h 6, 12 y 24 h. Después de eso, se midió el número de células, que estaban en cuatro fases de mitosis (profase, metafase, anafase y telofase) e interfase (el período de preparación celular para la división[98]).
En promedio, la mitosis dura 1 a 2 h e incluye la separación del material duplicado preliminar, la división del núcleo, la división de la propia célula y la citocinesis. Los datos obtenidos demostraron que el número inusualmente grande de células estaba en la profase y metafase, pero especialmente en la metafase. Si la tasa de replicación del ADN se retrasa solo durante la profase (el comienzo de la formación del huso de división, el sistema de cadenas de proteínas), la radiación de ionización podría romper las cadenas cuando son sensibles al daño. Sin embargo, para verificar esta influencia, es necesario realizar experimentos en profundidad sin la presencia de deuterio en moléculas de ADN y enzimas para determinar si la replicación del ADN se acelera en este caso en comparación con el ADN que contiene concentraciones habituales de deuterio.
4.19 Enlaces de hidrógeno y deuterio en el ADN
Como ya se ha señalado, la molécula de ADN es una doble hélice torcida a la derecha que consta de la estructura de azúcar-fosfato y compuestos de nitrógeno (Fig. 4.10). Registra toda la información sobre un organismo vivo específico en forma de fragmentos cíclicos alternos que contienen grupos carbonilo y amina. Hay cuatro tipos de tales fragmentos: adenina, timina, guanina y citosina (A, T, G y C). Están dispuestos en forma de colgantes laterales a lo largo de todo el modelo polimérico de ADN. El orden de alternancia de estos fragmentos determina la individualidad de cada criatura viva.
En una interacción pareada (A – T y G – C), aparece el enlace de hidrógeno entre ellos. Es este enlace el que sostiene dos moléculas de ADN en forma de doble hélice ampliamente conocida. Se cree que el deuterio afecta los procesos biológicos al alterar la formación de enlaces de hidrógeno, ya que los enlaces formados por átomos de deuterio son más fuertes que los formados por el átomo habitual de hidrógeno[99]. Enlace de hidrógeno y ADN En la molécula de ADN, el enlace de hidrógeno G – C y A – C, que se forma entre las hebras de la doble hélice, presenta interés. Es difícil determinar un valor exacto de la fuerza del enlace de deuterio. La fuerza de los enlaces de hidrógeno individuales del ADN fue evaluada; sin embargo, se cuestiona la precisión de su modelo. Se cree que los enlaces deuterio en las enzimas que afectan al ADN son normalmente entre 0,4 y 1,7 kJ/mol más fuertes que el enlace de hidrógeno habitual. Esto puede evaluarse mediante diferentes métodos que requieren un cálculo considerable.
Las tablas 4.2 y 4.3 proporcionan los datos que reflejan el cambio en la fuerza del enlace de hidrógeno en presencia de átomos de deuterio en los enlaces de hidrógeno. Como puede verse en los datos de la tabla 4.2, el reemplazo de algunos átomos de hidrógeno por átomos de deuterio afecta fuertemente la fuerza del enlace de hidrógeno, y en otros casos el efecto, que es más sustancial y máximo, se puede observar cuando todos los átomos de hidrógeno se reemplazan con átomos de deuterio.
Tabla 4.2 Fuerzo del enlace de hidrogeno y el formado por un átomo de deuterio en compuestos de hidrógeno.
 12.14.34.png)
La longitud del enlace de hidrógeno en la replicación y reparación del ADN, la forma de las moléculas enzimáticas que controlan estos procesos es extremadamente importante. El deuterio reduce algo la longitud del enlace e inhibe el funcionamiento apropiado de las enzimas. Sin embargo, lo más probable es que este efecto sea bastante insuficiente, posiblemente del orden de ~ 1%. En términos generales, la formación del enlace de hidrógeno en la molécula de ADN es un proceso de cooperación que afecta la interacción de apilamiento e involucra a toda la molécula. Si se utilizan enzimas como conductores, la fuerza del enlace deuterio en la molécula de ADN es 0.5 a 2.0% mayor que en un enlace de hidrógeno ordinario. Cuando existe una pequeña concentración de deuterio en la naturaleza, y hay un pequeño aumento de la fuerza del enlace de deuterio en comparación con el enlace de hidrógeno habitual, es tentador concluir que el deuterio no produce ningún impacto negativo sustancial en el ADN a la concentración de 155 ??m. Sin embargo, el fortalecimiento del hidrógeno deuterio también produce otros efectos potencialmente dañinos.
Mecanismos probables de la influencia nociva del ADN. Enzimas deuterizadas según una teoría bien conocida, el deuterio afecta negativamente la forma de las moléculas enzimáticas involucradas en los procesos del ADN. Este concepto principal fue propuesto por T.G. Griffith en el artículo "El posible papel del deuterio en la estimulación del envejecimiento y otros mecanismos y procesos biológicos[100]". Cuando se incluye deuterio en una reacción química, es necesario considerar pequeños cambios en el efecto de inducción porque el deuterio es más permeable a la electricidad que el hidrógeno. El efecto hiperconjugable se ha hecho evidente desde que ?D3, por ejemplo, está menos deslocalizado que ?H3 y, lo que es más importante, la longitud efectiva del enlace C–H se acorta, lo que afecta los efectos estéricos. Esto confirma la opinión de que para cualquier molécula estereoespecífica de la enzima que contenga destrona (núcleo de deuterio) en la sección importante, el potencial estará involucrado en reacciones erróneas.
Tabla 4.3 Reforzamiento del enlace D en comparación con un enlace típico H -según tabla 2.3%
 12.16.11.png)
4.20 Interferencia del deuterio en la reparación del ADN con enzimas
Un gran grupo de enzimas y proteínas participan en la replicación y reparación del ADN. Algunas de estas enzimas utilizan activamente enlaces de hidrógeno externos. Son potencialmente muy sensibles a los efectos negativos del deuterio disponible. Una proteína muy importante, la p53, es responsable de la reparación del ADN. Varios tipos diferentes de daños en el ADN pueden activar p53. Por ejemplo, las rupturas de dos cadenas de ADN después de la radiación gamma, la aparición de rupturas de doble cadena de ADN causadas por la radiación gamma, la aparición de intermediarios de reparación de ADN después de la radiación UV o el daño químico del ADN son todos los medios de activación de p53. Cabe señalar que más del 50% de las enfermedades humanas van acompañadas de una mutación genética que forma la proteína p53[101].
4.21 Retraso de la replicación del ADN
El deuterio también puede inhibir la enzima DnaB, que es responsable de desplegar y separar el ADN durante la replicación. Otras enzimas, como primasa y polimerasa, juegan un papel importante en la síntesis de ARN y la unión de nucleótidos a la cadena de ADN en el curso de la replicación. Si se suprime la acción de una de estas enzimas, la tasa de replicación del ADN también se puede suprimir notablemente, y es entonces cuando el ADN es más vulnerable al daño por radiación.
Por tanto, el deuterio puede servir como catalizador para la degradación del ADN en un nivel natural de exposición a la radiación. La inhibición de las secciones de enlace en la replicación del ADN. El proceso de replicación del ADN es, en cierta medida, como la reparación de una ruptura de doble hebra del ADN. El complejo de enzimas permite identificar y restaurar las hebras de ADN ubicadas en ciertas secciones de enlace. Los enlaces de hidrógeno a menudo actúan sobre estas secciones. Si hay deuterio sobre ellos, los efectos sintéticos y la mayor fuerza de los enlaces pueden inhibir la reparación del ADN. A su vez, si la tasa de reparación del ADN disminuye sustancialmente, la radiación ionizante puede continuar impidiendo el proceso de reparación. Aquí es cuando la hebra se destruye y aumenta la sensibilidad al daño por radiación adicional.
La eficacia del uso del agua empobrecida en deuterio según los datos[102], el uso de agua ligera se diferencia de varios antioxidantes, estimulantes de HGH, vitaminas y otros medios de antienvejecimiento en un aspecto clave: el agua ligera no cambia composición química en asimilación. El agua ligera consumida presenta efectos directos a nivel celular. Sin embargo, para que estos efectos se noten, es necesario eliminar el contenido de deuterio. Mediante reacciones exotérmicas que sustituyen al deuterio, los átomos son reemplazados gradualmente por átomos normales de protio. Este proceso ocurre más rápido cuando la concentración de deuterio en el agua potable está en el nivel más bajo posible. El deuterio ingresa constantemente al organismo, que consume los alimentos que se cultivan y cocinan con agua ligera. El agua ligera puede proteger el ADN del daño y ayudar a los mecanismos de reparación del ADN; sin embargo, por sí sola, el agua ligera no restaurará directamente el ADN. Por lo tanto, queda la pregunta de si el agua ligera puede “rejuvenecer” los organismos y ayudarlos a funcionar de manera más eficaz.
4.22 Conglomerados de agua (clusters)
4.22.1 Estructuras de agua, propiedades y formación de conglomerados
Recientemente, se ha prestado mucha atención a la formación de grupos en el agua. Esta formación está determinada por la participación de moléculas de agua en la formación de enlaces de hidrógeno intermoleculares[103]. Los defensores de la interpretación clásica de la estructura de los líquidos creen que el ciclo de vida de los grupos de agua es muy corto (del orden de 10-12); como resultado, un grupo de formaciones relucientes con una vida útil corta no puede afectar de manera visible las propiedades del agua. Los partidarios de la idea de la estructuración del agua implican que los racimos en el agua pueden conservarse durante mucho tiempo; por lo tanto, no se puede ignorar la presencia de asociados en varios casos.
Creemos que las propiedades fisicoquímicas del agua están completamente determinadas por el estado estructural del agua, que depende de su composición de racimos (Fig. 4.14) y de factores tales como el tamaño del racimo y la concentración de los racimos. La estructura del agua líquida ha sido objeto de una extensa investigación[104]. El estudio de la estructura del agua en el siglo XX dio lugar a numerosos conceptos, siendo el más importante el reconocimiento de la presencia de dominios ordenados.
Estas teorías (los defectos estructurales de Samoilov, las estructuras de Bernard-Fowler, los "hidratos" de Poling, los brillantes grupos de Frank y Wien y muchas otras) se discutieron intensamente en varios estudios[105]: (1) El orden remoto no se detectó ni auténtica ni experimentalmente en el agua; (2) se estimó que la vida útil de los conglomerados no superaba los 10–10–10–9s; 3) hay razones para creer que la presencia de procesos cooperativos en el agua está relacionada con la transferencia de los defectos de orientación L y D de Bierumme[106] .
 19.43.03.png)
Fig. 4.14 Posibles agrupaciones de agua
Los métodos de dispersión de rayos X o de dispersión de neutrones no confirmaron la formación de estructuras ordenadas con dimensiones superiores a las unidades de nanómetros[107]. Esto, a su vez, no concuerda con los datos sobre anomalías de histéresis de temperatura del indicador de refracción del agua[108] o dispersión anómala de agua en soluciones acuosas de compuestos orgánicos[109]. Por lo general, los cálculos teóricos de los grupos de agua se limitan a decenas de moléculas o cerca del límite de fase. M. Chaplin, por ejemplo, ha calculado y propuesto un modelo de agua cuya base es el icosaedro (Fig. 4.15). Dado que una capacidad muy pronunciada de autoorganización debido a la formación de enlaces de hidrógeno es característica del agua, propiedades del agua como, por ejemplo, la fluidez y la capacidad de formar hidratos de gas implican la formación de estructuras ordenadas expandidas como el agua continua. modelo[110]. Los datos de emisión acústica de soluciones acuosas en la región de 0.5 Hz a 2.5 kHz implican la presencia de una fuente generadora de ondas sonoras con una dimensión lineal mayor que las dimensiones de los grupos moleculares[111].
 19.47.28.png)
Fig. 4.15 Agua de icosaedro gigante
En la actualidad se ha reconocido que, debido a la existencia de enlaces de hidrógeno intermoleculares, las propiedades del agua difieren marcadamente de las propiedades de otros hidruros. Por ejemplo, tienen una estructura asociativa[112], cuya presencia a escala global determina el clima de la Tierra, así como el origen y soporte de la vida. Las propiedades anómalas del agua, al igual que las propiedades anómalas de las moléculas de oxígeno, son los principales factores que determinan la existencia de la biosfera terrestre. Las propiedades únicas e inusuales del agua en los estados líquido, sólido y gaseoso se deben a la presencia de enlaces fuertes entre las moléculas de hidrógeno: dos enlaces H en los que la molécula ?2? es un donante de protones y dos H- enlaces con la participación de un par solitario de electrones del átomo de oxígeno. Una estructura tan básica como la de la organización del agua[113] se ha adoptado como base para la consideración de modelos estructurales y propiedades del agua. Los enlaces H determinan la formación de estructuras supramoleculares bastante estables: grupos, nano y microdominios, cuyos valores dependen de la presencia y parámetros de interfaces con un cuerpo sólido, gas u otro líquido.
Muchas propiedades anómalas del agua hasta hoy no se han explicado por completo. Una de las razones de esto, desde nuestra perspectiva, es que los investigadores han ignorado en gran medida el fenómeno de la formación de grupos de agua. Una propiedad anómala del agua es que exhibe una gran capacidad de autoorganización debido a la formación de enlaces de hidrógeno y efectos cooperativos[114].
Por tanto, las estructuras primarias pueden ser dímeros, trímeros y oligómeros. Las estructuras cíclicas con las cinco y seis moléculas de agua forman varios poliedros. El más importante de estos poliedros tiene un eje de simetría de quinto orden (dodecaedro e icosaedro). Estos poliedros son importantes porque pueden ayudar a explicar muchas de las propiedades ilusorias del agua; proporcionan información, por ejemplo, sobre la volatilidad del agua y su capacidad para formar hidratos de gas.
 19.48.52.png)
Fig. 4.16 Representación esquemática de 512 grupos dodecaédricos (H2O) 20 (a), icosaédricos homológicos (H2O) 100 (b) y (H2O) 280 (c) grupos.
A partir de tales elementos estructurales, se pueden formar hebras largas y estructuras especiales que llenan el volumen. El movimiento de moléculas pequeñas como el agua en estado líquido a temperatura ambiente, de acuerdo con las nociones contemporáneas, tiene la naturaleza de fluctuaciones giratorias cerca de la posición de equilibrio, lo que puede conducir a la formación de grupos. Solo con un aumento de la temperatura del agua a 70-80 ° C, de acuerdo con la relación de Einstein 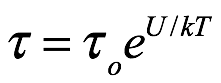 , donde τ es el tiempo de correlación del movimiento rotatorio) el movimiento de las moléculas se asemeja al movimiento en la fase gaseosa, reduciendo sustancialmente la estabilidad de los racimos. Dado que la reducción de la temperatura actúa sobre la estructura del agua como factor de ordenamiento, es posible la formación de racimos a partir de las moléculas de dímeros, trímeros y asociados con un gran número de moléculas. Dependiendo del método de investigación, se pueden observar desde 2–15 hasta 50–60 y más moléculas de agua[115]. En nuestra opinión, tales resultados son evidencia del hecho de que la gama de tamaños existentes es bastante amplia. Recientemente, la idea de formación de conglomerados ha recibido un reconocimiento y una confirmación experimental cada vez más amplios a través de diferentes métodos[116].
, donde τ es el tiempo de correlación del movimiento rotatorio) el movimiento de las moléculas se asemeja al movimiento en la fase gaseosa, reduciendo sustancialmente la estabilidad de los racimos. Dado que la reducción de la temperatura actúa sobre la estructura del agua como factor de ordenamiento, es posible la formación de racimos a partir de las moléculas de dímeros, trímeros y asociados con un gran número de moléculas. Dependiendo del método de investigación, se pueden observar desde 2–15 hasta 50–60 y más moléculas de agua[115]. En nuestra opinión, tales resultados son evidencia del hecho de que la gama de tamaños existentes es bastante amplia. Recientemente, la idea de formación de conglomerados ha recibido un reconocimiento y una confirmación experimental cada vez más amplios a través de diferentes métodos[116].
Debido al rápido desarrollo de poderosas tecnologías ab initio, se obtuvieron las respuestas para la mayoría de las preguntas fundamentales relacionadas con la naturaleza de la formación de enlaces de hidrógeno y la estructura de los grupos de agua[117]. Por lo tanto, con el uso del método ab initio MP2 se calcularon las estructuras geométricas y electrónicas de los complejos de agua con ozono[118]. También se determinaron las frecuencias vibratorias armónicas y las constantes rotacionales H2O, HDO y D2?. El estudio de diversas estructuras de hidratos de gas nos brinda la posibilidad de ver el cúmulo icosaédrico. La investigación de la estructura de los grupos de agua ha dado como resultado la identificación de hasta 280 moléculas de agua (Fig. 4.16). También se han establecido las características estructurales y de orientación de varias moléculas "huésped" en el volumen central del grupo icosaédrico. Se ha demostrado que los grupos de monómeros de agua tienen la mayor energía de estabilización. El método de dinámica molecular[119] permite a los investigadores calcular las propiedades termodinámicas de los grupos de agua pura, así como los agregados de agua con inclusión de las moléculas de CO o CO2. Para la investigación de la estructura de los grupos de enlaces H en metanol y agua cerca de la curva de saturación y el punto crítico, se utilizó el método de dinámica molecular[120]. Los métodos de Monte-Carlo de modelado matemático para grupos de hidratos con un ion se utilizaron al estudiar la influencia de la radiación electromagnética de baja intensidad en los grupos de agua con presencia de iones de sodio y potasio[121]; presenta los resultados de una investigación experimental y teórica sobre la interrelación entre la estructura y las propiedades eléctricas de los clusters.
Se observaron grupos que contienen seis, siete, ocho y nueve moléculas de agua en la Nanotecnología Centre en Londres y la Universidad de Leibniz en Hannover durante una investigación de las propiedades del hielo a nivel nano[122]. Además de los experimentos, se utilizó enfoques teóricos de la mecánica cuántica. El enfoque complejo mostró que, a diferencia del hielo cristalino, donde la energía de unión es idéntica entre todas las moléculas de agua, en los nanoclusters, los enlaces fuertes y débiles (a las distancias correspondientes) alternan entre moléculas individuales.
Los grupos de agua atrajeron mucho interés porque se forman precisamente dentro de un intervalo de temperatura natural y son capaces de producir variaciones locales “suaves” en las propiedades del agua, ya sea impulsando o ralentizando los procesos biológicos y otros procesos naturales. Al considerar el agua como catalizador de procesos bioquímicos, se debe prestar especial atención a los resultados de la investigación que tratan de la formación de racimos de agua. Creemos que el estudio de los mecanismos de catálisis de clústeres basado en los enfoques fundamentales del proceso de catálisis[123] abrirá procesos de agua hasta ahora desconocidos.
4.22.2 Conceptualización de capas límite de clústeres
El concepto, que se está desarrollando actualmente, se refiere a las capas límite de los grupos de agua; el límite de fase en el agua (independientemente del origen de la estructura del modelo de conglomerados) es necesario para explicar el vínculo entre la estructura del agua y sus propiedades. Tales propiedades incluyen, por ejemplo, la capacidad de sobreenfriamiento y la redistribución de impurezas como deuterio, gases disueltos y SAS.
Las propiedades de la solución y los sistemas dispersos también son importantes. El uso del modelo de agua como un líquido de racimo, en el que moléculas con una estructura variable de Hbonds actúan como capa límite, permite describir cuantitativamente muchos fenómenos, como el del sobreenfriamiento. La acumulación de impurezas, que compensan las cargas de polarización que ocurren simultáneamente. La concentración de sustancias de la capa superficial que están menos polarizadas que ?2? dará como resultado el blindaje de las cargas de los grupos y, en consecuencia, el debilitamiento de los enlaces de hidrógeno ubicados fuera de los grupos. En consecuencia, la concentración de la capa superficial dará como resultado la estabilización de la estructura del grupo. En relación con varios experimentos llevados a cabo con éxito sobre el estudio de los racimos gigantes de agua heterofase (GHWC[124]), la determinación de los racimos como fragmentos de agua con las propiedades variadas requirió la perfección de la terminología. Esto hizo posible diferenciar los grupos de acuerdo con el número de moléculas de agua que contenían. Los GHWC, o "microvolúmenes" de agua, involucran moléculas de HDO en su formación[125]. La existencia de GHWC permite la diferenciación de asociados ordenados de tamaños más pequeños, como microrracimos (que contienen de 2 a 20 moléculas) o pequeños grupos (de 20 a 80 a 100 moléculas). Los grupos de heterofase gigantescos contienen 010-1013 moléculas de agua.
La fuerza de movimiento de la formación de cúmulos, según Frenckel[126], es la orientación y polarización de las moléculas de dipolo líquido. Para el agua, que está en la mayor parte de la muestra, las fuerzas ordenadas primarias son estructuralmente similares a las secciones de las paredes de un recipiente de vidrio. Además, de acuerdo con las nociones sobre GHWC que hemos desarrollado, la formación de racimos es promovida por el deuterio disuelto.
Según uno de los mecanismos propuestos, el papel del deuterio implica la formación de la red volumétrica formada por cadenas que contienen HDO. La capacidad de los óxidos para formar grupos superficiales ácidos y básicos con la participación de los centros de Lewis y Brensted[127] depende del efecto ordenado de las superficies relacionadas estructuralmente, por ejemplo, el silicato. La afinidad de los centros de la superficie del óxido con los pares de electrones o con los protones da como resultado la adsorción de moléculas de agua en una determinada configuración. Por lo general, esta configuración de cargas superficiales corresponde a la estructura del hielo de los óxidos de silicona. Tras la adsorción de agua, las policapas cercanas a la superficie del silicato actúan como factor de nucleación y forman pequeños grupos de agua. En este caso, la impureza de HDO en el agua puede jugar un papel decisivo en la formación y estabilización de GHWC. La energía de hidratación iónica del electrolito excede la energía del enlace de hidrógeno del agua en varias veces. En este sentido, al añadir las primeras porciones del electrolito al agua, debido a la entrada de un HDO para hidratar las capas de iones, es posible que se formen nuevos racimos y se produzcan algunos cambios en la estructura del GHWC.
El agua varía en la estructura, cantidad y composición de micro racimos y pequeños racimos bajo el efecto polarizador de los iones del electrolito disuelto en el agua, que está en contacto con la superficie del silicato. Los investigadores asumieron la posibilidad de destruir enlaces de hidrógeno cooperativos introducidos por el electrolito[128]. En nuestra estimación, la existencia de enlaces H cooperativos que son capaces de abarcar todo el volumen de la muestra de agua, en combinación con el efecto polarizador de la superficie del silicato, apunta directamente a la posibilidad de la formación de conglomerados de gran tamaño, como GHWC.
Se escribió los métodos de obtención de racimos enriquecidos con agua. Así, utilizaron la energía de cavitación de las burbujas de aire para obtener agua que contenía microrracimos[129]. El proceso se repitió con un aumento de la temperatura del agua a 60 °C. Creemos que la rotura de las burbujas de cavitación va acompañada de la destrucción de las estructuras moleculares del líquido a estructuras más pequeñas, lo que da como resultado la formación de microclusters. Los racimos de agua fueron producidos por el tratamiento magnetohidrodinámico[130]. La observación directa de grupos es difícil; por tanto, al estudiar las propiedades del agua (RMN, IR y espectros UV de interferencia del rayo láser, Raman, viscosidad, electroconductividad, etc.), sus anomalías se refieren a las manifestaciones de cúmulos. El agua inicial se hirvió y se convirtió en vapor antes de pasar a través de un campo magnético. Luego se condensó a una temperatura un poco superior a 0 ° C, irradiando en la región del espectro IR al UV. En este caso, se añadió metasilicato de sodio <1% al condensado como plantilla de cristalización. De esta forma se obtuvieron los microcristales de agua. La presencia de racimos en el agua se vinculó con un ancho de la señal de RMN, viscosidad y conductancia eléctrica. Su dimensión se evaluó por el ancho de la línea de RMN al nucleido de oxígeno 17O. Para ello, el agua investigada se vertió en una placa de laboratorio de cuarzo de 2 cm3 de volumen, colocada en el resonador del espectrómetro de RMN. Los microrracimos de agua envían una señal de RMN <115 Hz de ancho. La conducción eléctrica de los grupos de agua constituía ~ 3.7 μS / cm, con una tensión superficial de menos de 61 mH/m. La concentración de microrracimos que contienen de 3 a 15 moléculas de agua se evaluó mediante la amplitud y banda ancha de la señal de RMN, utilizando el software. Un error sustancial por este método puede introducir efectos magnetohidrodinámicos.
4.22.3 Influencia de los isótopos de hidrógeno en la agrupación: pequeños grupos de agua
Los efectos isotópicos sobre el agua pesada altamente concentrada no existen en condiciones naturales. En la actualidad, estos efectos se han estudiado bastante bien. Sin embargo, el impacto de las impurezas sobre las moléculas pesadas de agua, principalmente el deuterio, y sobre la estructura y propiedades del agua es una situación completamente diferente. Los resultados obtenidos al investigar el papel del deuterio en la formación de GHWC son de especial interés ya que no solo amplían el campo de conocimiento sobre la estructura del agua sino que también propician el desarrollo de hipótesis sobre la implicación del agua en el origen de vida[131].
 20.26.42.png)
Fig. 4.17 Cadenas de HDO en agua: (a) con un enlace de oxígeno H; (c) una cadena reforzada por la participación de la interacción del deuterio con las moléculas de agua vecinas; (b, d) Imag4 esquemáticas.
El deuterio en agua está completamente unido a HDO. Dado que el ion D+ está unido a un átomo de oxígeno más fuerte que el ?+ (la movilidad del ion D+ constituye 0,7 de la movilidad del ion ?+) en una molécula HDO, los enlaces intermoleculares de deuterio de la molécula HDO no se expresan como fuertemente como enlaces de hidrógeno de la molécula ?2?.
A las concentraciones habituales, la impureza de HDO, que posee una tensión superficial baja, puede considerarse SAS, cuya localización en la capa límite estabiliza la estructura del grupo. La no equivalencia de los enlaces N y D intermoleculares determina principalmente una posible orientación de las moléculas de HDO vecinas en el agua. Además, la influencia de las fuerzas de orientación y la polarización de la migración dan como resultado la formación de cadenas asociadas a partir de moléculas de HDO. La figura 4.17a muestra las cadenas más simples de moléculas de HDO en el agua; también presenta una formación más compleja y estable (ver Fig. 4.17c) de las moléculas HDO y moléculas vecinas ?2?.
Las condiciones para la formación de la red energéticamente estable de los enlaces de hidrógeno, pueden realizarse en el agua mediante la formación de cadenas y una red volumétrica de moléculas de HDO. En la figura 4.18 se ilustran fragmentos de modelo de una red plana construida con moléculas de HDO en agua. De manera similar, las moléculas de HDO forman una red tridimensional en la mayor parte del agua. Con un aumento en los niveles de concentración de deuterio, aumenta el número de fragmentos de la red de clústeres, mientras que la dimensión de las mallas de la red disminuye. En el agua empobrecida en deuterio, la integridad de la red volumétrica se ve sustancialmente alterada, debido a la deficiencia de las moléculas de HDO para construir cadenas. Hasta el día de hoy, no está claro cómo los cambios en las propiedades fisicoquímicas del agua están sujetos al drástico debilitamiento del deuterio, es decir, bajo la condición de que no se pueda formar la red de HDO (ver Fig. 4.18c).
 20.27.29.png)
Fig. 3.6 Un fragmento de red de las moléculas de HDO (puntos rojos), lleno de moléculas de H2O: (a, b) es el contenido aumentado de deuterio en el agua y el contenido habitual de deuterio respectivamente; (c) agua fuertemente empobrecida por el contenido de deuterio.
 20.31.13.png)
Fig. 4.18 Ley de Bouguer-Lambert-Beer (vc: obsc) para la dispersión de la luz láser GHWC en el agua: ? agua bidestilada; ? – D — soluciones (? — NaCl (~ 0,001 M), C — MgCl2 (~ 0,001 ?), D — ZnCl2 (~ 0,001 ?); —agua bidestilada (los indicadores de dispersión se determinaron cuando se utilizó como fondo de agua (4 ° ?); F, 1 — agua del grifo (centro de Moscú); 2, 3 soluciones de HCl (pH <4); 4— agua pesada; 5 agua con protio; 6 — agua de mar (el Mar Negro); 7-9 fresco subterráneo embotellado comercialmente (Aqua Minerale, Bon Aqua, Svyatoi Istochnik); solución 10 — NaOH (~ 0,001 M).
Las investigaciones han demostrado que se forman al menos dos estructuras en el agua. Esto fue corroborado por los datos del experimento de la figura 4.18, que ilustra la ley de Bouguer-Lambert-Beer para la dispersión de luz láser GHWC de agua diferente. En la figura se puede ver que efectivamente todas las muestras de agua analizadas en términos de su estado estructural pueden clasificarse en dos tipos.
Debido a la falta de uniformidad de los enlaces de hidrógeno y deuterio, las cadenas de las moléculas HDO y H2O son más estables cuando las moléculas se empaquetan en forma de formaciones helicoidales. Su composición se vuelve más compleja y las posibilidades funcionales aumentan a medida que aumenta la adsorción selectiva de impurezas. Según el modelo GHWC, en el que el HDO localizado estabiliza la superficie del racimo, es necesario aclarar la proporción de deuterio y protio en el agua. En estas circunstancias, las moléculas de HDO son suficientes para provocar la formación de una película que cubra toda la superficie del grupo. Los cálculos han demostrado que las proporciones de moléculas de agua en la mayor parte del grupo y en su superficie (NV / NS) están vinculadas con las dimensiones lineales del grupo (α) y las partículas (d) que lo forman de la siguiente manera:
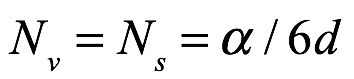 (3.1)
(3.1)
De la fórmula, se deduce que, a medida que disminuye la dimensión del conglomerado, la relación NV/NS disminuye. Al llenar la capa superficial, por ejemplo, aumenta la cantidad relativa de moléculas. En referencia al caso más simple (para un asociado cúbico), bajo la condición d = 0.2 nm y de acuerdo con la fórmula (3.1), se calculó lo siguiente: para ubicar las moléculas de HDO en la monocapa superficial del clúster y mantener la relación de protio y deuterio 1/6.000, las dimensiones del racimo deben ser de al menos 7.2 μm. Cabe señalar que la descripción proporcionada es puramente geométrica y no se aplica al aspecto físico del proceso. En otras palabras, no se puede generalizar como la razón de la localización de agua pesada en la capa superficial de los grupos de agua. Para cumplir la proporción en el agua deuterio: protio = 1: 6.500 en la superficie de una sola malla, la red volumétrica debe ser ~ 4.8 × 109 de las moléculas de HDO, y en la mayor parte de una sola malla, aproximadamente 3.2 × 1013 de las moléculas de ?2?.
La estructura del agua dentro de una malla de la red de las moléculas de HDO puede describirse mediante un modelo de dos estructuras que permite la formación de pequeños grupos de agua. Así, como se sabe[132], en una molécula de agua “híbrida”, donde un átomo de hidrógeno es sustituido por deuterio, el enlace de hidrógeno se forma a partir del átomo de deuterio.
Una excepción es el H5O2 ±, donde el enlace de hidrógeno es especialmente corto. Por tanto, desde nuestra perspectiva, la estructura del agua depende directamente del contenido de deuterio. Desafortunadamente, no se han obtenido datos suficientes para una determinación confiable de la estructura de pequeños conglomerados. De hecho, las muestras experimentales contienen varios isómeros y, por lo tanto, los grupos que tienen diferentes dimensiones a menudo son muy difíciles de separar. En los últimos años, se han propuesto cientos de modelos para la formación de conglomerados de agua (ver Fig. 4.1). A pesar de esto, los cálculos de las propiedades del agua siguen siendo en gran parte inexactos.
Un simple cálculo es suficiente para determinar que 1 dm3 de agua contiene 125 g de hidrógeno, de los cuales 43 mg son átomos de deuterio. Esto nos proporciona el fundamento para juzgar la presencia de una concentración sustancial de deuterio, que no puede ignorarse en los cálculos. En consecuencia, en las moléculas de agua pesada donde ambos átomos de hidrógeno están sustituidos con deuterio y tritio (D2O y T2O), el enlace de hidrógeno es más fuerte que en el agua ligera (H2O). También está muy estructurado; esto se confirma por los grandes volúmenes molares[133].
El efecto del isótopo de hidrógeno se ha considerado utilizando el ejemplo de oscilaciones simétricas y asimétricas del enlace OH en las series ???, HOD, DOD, (???)2, (DOD)2, DOD HOH, HOH – DOD, HOD – HOH. Se ha prestado especial atención al efecto cooperativo del agua, que fue postulado por primera vez por G.S. Frank y W. Wien[134]. De acuerdo con este efecto, la formación de un enlace de hidrógeno conduce al crecimiento de grupos y, a la inversa, la escisión de un enlace facilita la destrucción de todo el complejo. Las estructuras de los grupos de agua se optimizaron en el nivel teórico de segundo orden de Møller-Plesset (MP2). El conjunto base 6–311 ++G(d,p) se eligió tanto para la optimización como para la determinación de frecuencias. Las frecuencias de vibración se calcularon sobre la base de una estructura optimizada mediante el método mencionado anteriormente. A continuación, se obtuvieron Imag4 gráficas de grupos de agua y visualización de espectros de infrarrojos utilizando el programa Molden[135]. La energía de formación de conglomerados o energía de estabilización ES y la energía del enlace de hidrógeno E? se calcularon utilizando las energías totales del estado de equilibrio de un cúmulo y el estado fundamental de una molécula de agua mediante el MP4 (SDQ) con un conjunto de bases mejorado 6-311 ++G (2d, 2p):
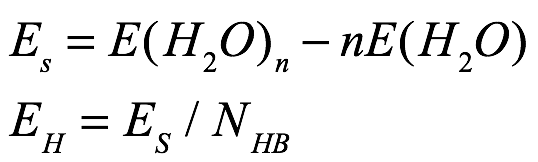 (3.2), (3.3)
(3.2), (3.3)
donde NHB: es el número de enlaces de hidrógeno en un grupo.
El parámetro del criterio de conversión para la energía y el gradiente del campo autoconsistente constituía 10−7. Monómero de agua (H2O) La longitud de enlace de ?– ? y el ángulo de valencia ??? en una molécula de agua optimizada por nosotros constituían 0.09595 nm y 103.47 ° respectivamente, lo que concuerda bien con los datos experimentales (0.09584 nm, 104.40[136] °). En una molécula de agua, se distinguen tres vibraciones diferentes: ν1: vibraciones simétricas; ν2: deformación del ángulo de valencia ???; vibración asimétrica ν3. Según los cálculos, los valores de estas frecuencias son iguales a 3.884,54, 1.628,61 y 400,77 cm−1; esto se puede comparar con las respectivas frecuencias vibratorias del monómero de agua observadas en las matrices del neón FTIR mediante un espectrómetro: 3.660,6; 1.595,6; 3.750,7 cm−1[137].
A diferencia del H2O, donde las oscilaciones vibratorias se combinan con el movimiento de ambos enlaces OH, en HDO las oscilaciones vibratorias ν1 y ν3 están asociadas solo con un enlace hidroxilo y constituyen: v1 = 2.864,3204, v2 = 1.427,2593, v3 = 3.946,26 cm−1.
Como resultado, un átomo de protio es más reactivo y se disocia fácilmente que el deuterio. Los datos del experimento indican el siguiente HDO: 2.722,9; 1.404,1; 3.699 µm−1. Las frecuencias vibratorias del espectro IR del monómero de agua pesada D2O son mucho más pequeñas, debido a un aumento en el peso atómico. Sin embargo, similar a ?2?, en oscilaciones asimétricas y simétricas están involucrados los siguientes modos: v1 = 2.802,03 (2.672,7), v2 = 1.191,21 (1.178,7), v3 = 2.931,60 (2.787) cm−1 (los paréntesis contienen datos experimentales de las frecuencias de oscilaciones armónicas de los isótopos del monómero de agua). Con base en el espectro de IR del monómero, se determinaron las constantes de rotación de los isótopos del monómero de agua y se dan en la Tabla 4.4.
Tabla 4.4 Constante rotacional del monómero de agua
 12.28.05.png)
Tabla 4.18 Parámetros geométricos optimizados del damero de agua
 12.29.29.png)
 20.43.27.png)
Fig. 4.19 El dímero de agua (H2O)2
Dímero de agua (H2O)2. El dímero de agua es su grupo más pequeño. Como resultado de la optimización, se encontró que el dímero se caracteriza por una estructura con un enlace de hidrógeno ordinario, perteneciente a la simetría Cs, donde se colocan ambos átomos de oxígeno y un átomo de la molécula de hidrógeno, el donante del enlace de hidrógeno. La longitud del enlace de hidrógeno en tal estructura es igual a 1.950 nm. Cabe señalar que el enlace de hidrógeno en la estructura del dímero de agua se desvía de la linealidad en 2.113 °, mientras que la distancia entre los átomos de oxígeno es de 0.2914 nm.
El átomo de hidrógeno del enlace O–H no participa en la formación de un enlace de hidrógeno y, por lo tanto, en lo sucesivo se denominará hidrógeno libre (?u). Un átomo de hidrógeno, por otro lado, está involucrado en la interacción como unido (?b). Según el cálculo, la energía de estabilización de un dímero de agua constituye 21,06 J/mol. Los datos obtenidos coinciden bien con dicha investigación experimental 22,2 ± 0,033 kJ / mol[138]. Los datos geométricos de la estructura del dímero optimizado, así como la forma del grupo de agua, se dan en la Tabla 4.4 y la Fig. 4.19.
Tabla 4.4. Frecuencia de oscilaciones armónicas de dímeros de agua
 12.35.08.png)
 20.45.53.png)
Fig. 4.19 Trímero de agua (H2O)3
Con base en los espectros IR, se determinaron las frecuencias vibratorias para (???)2, (DOD)2, DOD – HOH, HOH DOD, HOD – HOH, y se muestran en la Tabla 4.5. A partir de estos datos, se puede ver que los complejos en los que la formación de enlaces de hidrógeno involucra al deuterio tienen valores más bajos de frecuencias vibratorias. Esto es evidencia de un enlace de hidrógeno más fuerte.
El análisis de los espectros de IR obtenidos muestra que el pico de absorción en la frecuencia más alta aparece como resultado de la vibración asimétrica O–H de la molécula aceptora. El otro pico más bajo está asociado con la oscilación de la molécula donante O–H.
La tercera etapa de absorción es el resultado de la vibración simétrica O–H del modelo aceptor. Finalmente, la absorción a la frecuencia más baja corresponde a las vibraciones de estiramiento del grupo O–H unido de la molécula donante. Tal imagen está cualitativamente de acuerdo con los datos de la investigación experimental[139].
4.22.4 Trímero de agua (H2O)3
En cuanto al trímero de agua, en la actualidad continúa una discusión sobre la esencia de su conformación estructural. Algunos investigadores apuntan a una estructura en forma de cadena lineal abierta, mientras que otros creen que la estructura cíclica es mayormente estable[140].
El resultado de la optimización del trímero de agua es una evidencia de la estructura cíclica del grupo dado (Fig. 4.20). El trímero de agua optimizado es una estructura triangular equilátera con tres enlaces de hidrógeno. Los átomos libres de hidrógeno están dispuestos por encima y por debajo del plano formado por átomos de oxígeno. Debido a esta disposición, este complejo adquiere propiedades de compuestos quirales. Un trímero de agua es una estructura única; cada monómero de agua como parte de un trímero actúa como donante y aceptor del enlace de hidrógeno y también contiene un átomo de hidrógeno libre y uno unido. En comparación con el dímero, el trímero de agua tiene el enlace de hidrógeno más corto, cuya longitud promedio constituye 0.1926 nm.
La distancia de O–O (0.2797 nm) también disminuye, lo que resulta en el fortalecimiento del enlace de hidrógeno. La energía de la formación del trímero (64.25 kJ/mol) es el doble que la del dímero, ya que la disociación del dímero implica la rotura de un solo enlace de hidrógeno. Para que el trímero se descomponga en un monómero, primero es necesario destruir los tres enlaces de hidrógeno.
Tabla 4.6 Parámetros geométricos optimizados del trímero de agua
 12.37.34.png)
Los datos geométricos del estado de equilibrio del trímero se muestran en la Tabla 4.6.
4.22.5 Tetrámero de agua (H2O)4
El tetrámero del agua, al igual que su trímero, tiene una estructura cíclica, donde los átomos libres de hidrógeno se alternan sobre y debajo del lugar del anillo O–O–O–O formando una estructura con los elementos de la simetría S4[141]. Como el trímero, en el grupo del tetrámero cada monómero es simultáneamente un donante y un aceptor, pero, a diferencia del trímero, no poseen las propiedades de los enantiómeros ya que contienen un número par de átomos de oxígeno como parte del anillo. El tetrámero también conserva la tendencia exponencial a una disminución de la longitud del enlace ? – ? (0,2748 nm) y el fortalecimiento del enlace de hidrógeno (0.1791 nm).
Hemos obtenido dos estructuras optimizadas del tetrámero que se muestra en la figura 4.21. En una de las estructuras, los átomos de hidrógeno libres se alternan sucesivamente por encima y por debajo del plano del anillo ?4, y en la otra, los átomos libres se alternan en pares. La energía de formación de este último constituye 108.27 kJ/mol, que es 3,85 kJ / mol mayor que la energía de estabilización de la configuración alternativa. Los parámetros geométricos de estas estructuras se dan en las Tablas 4.7 y 4.21.
 20.56.21.png)
Fig. 4.21 Tetrámeros de agua (H2O) 4 I (a) y II (b)
Tabla 4.7 Parámetros geométricos optimizados del trímero de agua
 12.41.51.png)
Tabla 4.8 Parámetros geométricos optimizados de la forma alternativas de agua
 12.43.31.png)
4.22.6 Pentámero de agua (H2O)5
El pentámero de agua es uno de los grupos de agua más abundantes en la naturaleza. Se puede encontrar en la capa de disolvente de clatratos hidrófobos, moléculas de ADN y proteínas. En condiciones de laboratorio, los racimos de agua de pentámeros pueden obtenerse como resultado del paso de un gas inerte a través del agua en estado líquido, seguido de la expansión adiabática en el vacío[142]. La estructura del pentámero es como la estructura cíclica del trímero y también tiene propiedades quirales. La estructura estable del pentámero es como la estructura cíclica del trímero y también tiene propiedades quirales. El valor promedio de la longitud del enlace O–O en el pentámero, optimizado por nosotros, constituye 0.273 nm, y la desviación de la planaridad es 16.29 °. Hay cinco enlaces de hidrógeno en el grupo del pentámero, que coinciden al máximo con la línea ?–? (ángulo ?? – ?b = 3.5°). En el pentámero de agua, el efecto cooperativo se sigue conservando; por lo tanto, es más difícil romper el primer enlace de hidrógeno ya que el siguiente enlace se rompe más fácilmente que el anterior, y así sucesivamente. La energía de equilibrio del pentámero constituye 147.30 kJ / mol. Los datos geométricos y la figura de la estructura de equilibrio del pentámero se dan en la tabla 4.9 y en la figura 4.22.
Tabla 4.9 Parámetros geométricos optimizados de pentámetros del agua
 12.46.38.png)
Fig. 4.22 Pentámero de agua (H2O)6
4.22.7 Hexámero de agua (H2O)6
El hexámero de agua simboliza la transición de una estructura de racimo de dos a una tridimensional. Hemos optimizado una estructura de hexámero cíclico en forma de prisma (figura 4.23), donde cada monómero del agua está enlazado con tres enlaces de hidrógeno. Esto, (H2O)6 en total, representa 9 enlaces de hidrógeno[143]. En el hexámero, se pueden distinguir dos tipos de monómeros DDA (donante, donante y constituye donante y aceptor) y AAD (aceptor, aceptor y donante), cuya cantidad está equilibrada (3:3). Si miramos el hexámero desde un ángulo diferente, podemos ver que su cúmulo es nada menos que una superposición del dímero con un tetrámero, en el que los átomos de hidrógeno libres están dispuestos en pares sobre el plano del anillo ?4. La energía de estabilización del hexámero es la más alta entre los grupos considerados y constituye 183.96 kJ/mol.
 20.58.34.png)
Fig. 4.23 Hexámero de agua (H2O) 6
La otra estructura hexagonal optimizada por nosotros contiene solo 7 enlaces de hidrógeno y se asemeja a la forma de un "libro abierto" (Fig. 4.24).
Fig. 4.24 Isómero de agua O3 (H2O)
En este isómero, cuatro monómeros tienen un par DA; los otros dos están conectados por tres enlaces de hidrógeno y tienen la configuración DDA y AAD. Es obvio que el desequilibrio del tipo de monómero y una disminución en el número de enlaces de hidrógeno afectan el componente energético del grupo. De hecho, el isómero dado en 7,89 kJ / mol es menos estable que un prisma como análogo. Los datos geométricos para la estructura estable del hexámero se dan en la Tabla 4.10.
Table 4.10 Parámetros geométricos optimizados de hexámeros del agua
 12.51.12.png)
Los resultados de la investigación han demostrado que la estructura cíclica de grupos anillados es más beneficiosa en términos de energía que lineal. La disminución de la longitud del enlace ?-?, con un aumento en el tamaño del grupo, se correlaciona con una disminución de la longitud del enlace de hidrógeno, que se acompaña de un alargamiento de ?–?b. Una reducción de O–O da como resultado un aumento de la energía del enlace de hidrógeno (Tabla 4.25) en los grupos de agua (H2O) n para n = 2… 5.
Tabla 4.24 Energía de forma de conglomerados (Es) y energía de los enlaces de hidrógeno (EH)
 12.54.12.png)
La mayor desviación del enlace de hidrógeno de la linealidad se registró en el grupo de trímeros (21.3°). En el tetrámero, este indicador disminuye y alcanza una linealidad casi completa en el pentámero (3.5°). Por lo tanto, se deduce que los enlaces de hidrógeno en grupos tienden a alcanzar la linealidad y la estructura tetragonal es similar a la estructura del hielo.
Sobre la base del hexámero de agua, que debe considerarse como un compuesto dímero con el isómero tetrámero, se puede suponer que los grupos superiores, quizás, consisten completamente en unidades elementales (pequeños grupos de agua). Nuestros cálculos han confirmado el efecto cooperativo del agua bajo observación experimental. La energía de formación de racimos aumenta monótonamente con un aumento de su tamaño, de 21 en el dímero a 188 kJ/mol en el hexámero.
4.23 Interrelación de los efectos de la acción física sobre el agua y la agrupación
Habiendo analizado los últimos datos en la investigación de la estructura del agua, se pueden revelar los mecanismos y explicar los efectos únicos de los impactos físicos utilizados en los procesos de tratamiento y desinfección del agua como el tratamiento magnético, por ultrasonidos y ultravioleta.
El mecanismo del efecto del campo magnético en los sistemas de agua, el campo magnético permanente (MF) no debería afectar a los objetos diamagnéticos, como el agua. Al mismo tiempo, los resultados del uso práctico del tratamiento magnético son evidencia de la eficiencia de este método en el tratamiento del agua[144].
Hay varios puntos de vista que explican el impacto del tratamiento magnético preliminar en los procesos que ocurren en los sistemas de agua. Sin embargo, el mecanismo de acción de la MF en los sistemas de agua sigue siendo insuficientemente estudiado. La investigación experimental sobre magnetización de sistemas de agua[145] no es del todo reproducible y aún no proporciona una respuesta a la pregunta de qué factores están detrás de la acción del MF y si son capaces de causar cambios sustanciales en el agua misma y sus impurezas. En particular, se considera la cuestión del tratamiento magnético en relación con las nociones contemporáneas sobre la estructura del agua y las soluciones. Igualmente importantes son características del tratamiento de MF como el momento de la secuela de MF y el momento de la restauración del estado del sistema después de la finalización de su operación. Tales características dependen de la capacidad de los líquidos para lograr la relajación de sus propiedades. Se puede esperar que la fem de la celda de polarización electroquímica magnetohidrodinámica (MHD), como un rango de otras características de las soluciones[146] bajo la influencia de kT, disminuya con el tiempo t de acuerdo con la ley exponencial:
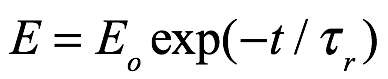 (3.4)
(3.4)
donde τr: el tiempo de relajación depende de la despolarización de los procesos del agua. Con base en la investigación realizada por el método de difracción de rayos X, dinámica molecular y Monte-Carlo, se demostró que la estructura tetraédrica del enlace de hidrógeno característico del agua en masa libre puede cambiar fuertemente en términos de efectos externos[147]. La capacidad del agua para conservar el estado característico de los efectos externos precedentes fue advertida por primera vez por biólogos y confirmada por métodos espectrales[148].
La relajación completa del estado depende de la posibilidad de procesos de rotación, traslación, orientación y movimiento migratorio de partículas individuales y sus agregados. El campo magnético en el líquido en movimiento afecta a todas las partículas cargadas simultáneamente y, por lo tanto, los gradientes de concentración de carga las partículas aparecen en toda la masa del agua. Después de que el líquido sale de la zona de influencia de MF, se forman los flujos de difusión, devolviendo el líquido a su estado inicial.
El tiempo de relajación τr de las moléculas libres de enlaces de hidrógeno cooperativos según la fórmula de Debye[149]
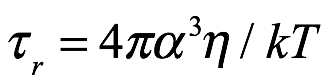 (3.5)
(3.5)
(donde α — es el radio de una partícula). Esta fórmula tiene el orden de 10−11 s, en particular para moléculas de agua (a la temperatura de 293 k, α = 0.193 nm, η = 10−3 Pa s) τr = 2.2 × 10−11 s. Sin embargo, si el agua contiene partículas más grandes, el tiempo de relajación puede medirse en minutos.
Relativo a la determinación experimental del tiempo de relajación, la fem se midió mediante diferentes intervalos de tiempo después de que el líquido saliera del campo magnético. En este caso, el líquido que fluye de la celda MHD colocada en el MF entró en la celda de manera similar a la celda colocada fuera del MF. Los electrodos de una celda se conectaron eléctricamente a los electrodos de la segunda celda solo a través del líquido que fluye. Los experimentos han demostrado que las diferencias de potencial en la celda de medición disminuyeron a medida que se retiraba de la celda MHD.
El tiempo de alcanzar la celda de medición por el líquido se calculó en base a su tasa. Más allá de la influencia de MF (en el caso dado, inducción de 0.8 T) de MHD, la fem disminuyó y después de 2 s tuvo un valor <10% de la medición inicial. Una fem MHD más larga en comparación con el valor esperado. Según los cálculos, la relajación de la fem de MHD es evidencia de la participación en el proceso de polarización de las partículas de MF, para las cuales la polarización de migración avanza a una velocidad mucho más lenta que para las partículas de dimensiones moleculares. En el agua y las soluciones observadas, tales partículas pueden ser grupos e iones que contienen capas de hidratos parciales, agua monomolecular y grupos de agua parcialmente destruidos.
Según los datos[150] de los artículos, la estructura continua del agua puede verse violada por la formación de grupos de al menos 4-6 a 300 moléculas. En condiciones normales, las fuerzas determinadas por kT y enlaces de hidrógeno del agua son proximales y, por lo tanto, los grupos a veces se describen como estructuras "relucientes" con una vida útil del orden de 10 ps. Por el contrario, las anomalías de las propiedades del agua, en particular el alto sobreenfriamiento, apuntan a los cúmulos como formaciones más estables.
La existencia de tales clusters se ha corroborado con métodos teóricos y numéricos[151]. Después de que se aplicó el tratamiento magnético a los procesos de reorientación y polarización de las moléculas de agua unidas en racimos, comenzó un período de relajación. Cálculos utilizados al mismo tiempo que la fórmula
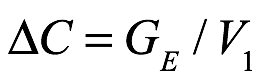 . (3.6)
. (3.6)
(donde Vl - es el volumen de líquido) demuestran los tiempos de relajación observables de la fem MHD (1… 2, 3s) que pueden lograrse mediante la despolarización de partículas con el tamaño de (7… 9) × 102 nm.
Durante mucho tiempo, las relajaciones del sistema nos han ayudado a explicar el proceso de tratamiento magnético en unidades que representan un imán permanente. Esta explicación incorpora la inserción de una tubería de material aislante en la tubería de agua. La eficiencia de unidades similares puede estar asociada a la presencia de un período más largo de relajación de la fem MHD, por lo que esta última actúa en el área magnética. Por ejemplo, a la velocidad del movimiento del líquido de 0.5 m/s, la fem residual actúa en el flujo de salida a una distancia de aproximadamente un metro.
La transformación de las propiedades del agua bajo el efecto del tratamiento electroquímico En la actualidad, debido a la simplicidad del hardware de fondo de la preparación "viva" (cátodo) y "muerta" (ánodo), el agua tratada electroquímicamente se esparce ampliamente. Detrás del método se encuentra la electrólisis del agua; sin embargo, los procesos que lo acompañan no han sido suficientemente estudiados hasta el día de hoy.
Un proceso en las zonas cercanas a los electrodos se agota no solo por las variaciones del pH sino también por el ablandamiento del agua del cátodo. Los experimentos han demostrado que después de 10 a 15 minutos de tratamiento en una unidad doméstica con un cátodo de acero, un ánodo de grafito con una densidad de corriente de 20 mA/cm2 y un gradiente de potencial de 50 V/cm, se forma peróxido de hidrógeno en el agua del ánodo. Esto se detectó mediante titulación directa de agua anódica con una solución de KMnO4, resonancia electrón-protón (EPR) y la aparición de una señal singlete de oxígeno atómico radical aniónico[152] ( ) en una muestra congelada de el agua del ánodo a la radiación UV.
) en una muestra congelada de el agua del ánodo a la radiación UV.
Condiciones para la formación de peróxido de hidrógeno a través de la reacción
 21.12.35.png)
en una celda electroquímica se realizan cerca del ánodo (provisto de la presencia de protones, la fuente de electrones y oxígeno). Los electrones se transfieren de los radicales libres (FR) o mediante el mecanismo de carrera de relevos de las moléculas de agua. La formación de peróxido de hidrógeno y el anión ?2?2– constituyen una etapa intermedia bien conocida en la liberación anódica de oxígeno[153]. En el ácido, el peróxido de hidrógeno del agua anódica actúa como oxidante responsable de la fuente del radical libre de oxígeno:
 21.12.39.png)
Bajo la influencia de los radicales libres, la destrucción o recombinación de los principales elementos del agua (??− , ?+, OH•, OH•_ , ?−, O•, H2O•, O2H•_ , ?3 ?+, O3H• etc.) es posible[154]. Los FR de vida corta catalizan procesos radicales mediante la transferencia en cadena de un electrón desapareado, lo que da como resultado la formación de cationes, aniones y moléculas neutras de FR, cuya entrada al organismo aumenta el consumo de antioxidantes.
Las impurezas orgánicas del agua son compuestos con fragmentos condensados policíclicos que contienen el electrón desapareado debido a los defectos de los enlaces intermoleculares. Estas impurezas se convierten fácilmente en radicales libres estables[155]. No es deseable que se incluyan en la bebida fragmentos que contienen radicales conocidos como FR estables (benceno, anilina, apoxílico, nitroxílico, aminílico, hidracílico, etc[156].). La sensibilidad de la estructura del agua a las inclusiones de partículas extrañas entre varias investigaciones termodinámicas[157] está bien documentada. Es probable que la red espacial de enlaces de hidrógeno, incluido el FR, inicie la formación de estructuras de agua anómalas.
El grado en que la electrólisis influye en el estado de la estructura del agua se investigó en la investigación actual sobre el agua tratada electroquímicamente mediante la sonda EPR, un radical estable cuyos espectros son sensibles a la variación del entorno local de la molécula.
Como resultado del tratamiento electroquímico del agua, las moléculas de dipolo adquieren la capacidad de orientarse al campo eléctrico externo, creando así la posibilidad de una estructura de agua ordenada. El notable orden molecular de moléculas no emparejadas puede observarse principalmente de acuerdo con el mecanismo de polarización de relajación.
Los dos efectos de la polarización de orientación de relajación de las moléculas de agua dan como resultado el orden gradual de la estructura del grupo; también conducen a la polarización de la migración del clúster, mientras que la estructura ordenada contribuye a un aumento de la microviscosidad en el tratamiento prolongado del agua. Los efectos a largo plazo del campo eléctrico incluyen la formación de pequeñas islas (grandes grupos); poseen una mayor viscosidad, debido a la orientación preferencial de los enlaces de hidrógeno a lo largo de las líneas eléctricas de campo.
El orden del sistema aumenta con el tiempo a través de la polarización de migración de grandes conglomerados, que se produce debido al aumento de su momento dipolar y polarizabilidad. De interés práctico es el período de conservación del efecto adquirido bajo la influencia del tratamiento electroquímico y, en particular, la conservación de las
estructuras con orientación cambiada.
Con base en los resultados obtenidos, es posible suponer que la naturaleza de la disminución en el tiempo de la correlación giratoria de la sonda después de la terminación del tratamiento del agua está determinada por la polarización de orientación de los grupos. En el caso del agua de cátodo, las dimensiones de los grupos constituyen (1,7… 1,8) × 104 nm, y para el agua de ánodo, 1,0 × 104 nm.
Absorción de sonido en el agua. Como ya se ha señalado, una de las razones de la difícil detección de los grupos de tamaño pequeño es el impacto de los métodos de investigación que deforma la estructura de agua más cercana. Es bien sabido que la ausencia de enlaces de hidrógeno en el agua podría resultar en una temperatura de fusión y ebullición mucho más baja que la conocida para el agua. Dentro del rango de temperatura del estado del agua líquida, la fuerza, los parámetros de ángulo y la cantidad de enlaces de hidrógeno varían. De hecho, el valor de la energía de los enlaces H del agua (18,8 kJ/mol) determina todos los procesos relacionados con la variación de su estructura más cercana. A temperaturas no superiores a ~80 °?, las formaciones estructurales en el agua se destruyen parcialmente. Consideremos, por ejemplo, el efecto de diferentes métodos de investigación sobre la estructura del agua.
Se puede suponer que la destrucción de las estructuras de los grupos está relacionada con los cambios de Qcl del orden del 10% de la energía de los enlaces de hidrógeno del agua (es decir, Qcl = 1,881 kJ/mol). Por lo tanto, el valor es bastante real para la temperatura ambiente, en la que las moléculas de agua se estructuran en agregados primarios similares al hielo o en grupos secundarios. En comparación, la Tabla 4.12 muestra las energías de radiación que afectan al agua en las investigaciones espectrales.
Tabla 4.12 Región del espectro y su energía de radiación correspondiente
 12.57.40.png)
Como puede verse a partir de estos datos, la mayoría de los métodos espectrales producen fuertes efectos en los objetos que se estudian. En el caso del agua, esto puede resultar en la destrucción de pequeños racimos o, para ser más exactos, en el parpadeo de sus estructuras y el cambio de sus formas y dimensiones.
Por ejemplo, el impacto de la radiación de rayos X supera el valor Qcl en 2 × 104veces. Las transiciones de los electrones de valencia correspondientes a tales energías se implementan independientemente de si las moléculas de agua están incluidas en formaciones supermoleculares. Esta condición también va acompañada de la destrucción de la estructura de agua cerrada y también de pequeños racimos. La radiación en la región visible del espectro supera Qcl en 80,4 veces.
Está claro que, si se ubicaran pequeños cúmulos en el agua, entonces en esa iluminación se produciría una reestructuración continua como resultado de la transformación de los electrones de valencia bajo los efectos de la luz. En los métodos combinados que utilizan fuentes de luz monocromáticas, la acción de la última puede reducirse mediante la selección de la frecuencia. La región de infrarrojos remota es aceptable en términos de efectos mínimos del método de investigación. En este caso, la radiación unitaria excede Qcl solo en 2.07 veces.
Los efectos más débiles se ejercen por métodos que utilizan ondas de radio cortas (RMN, EPR, DENR, etc.). Sin embargo, en estos métodos una muestra de agua, además de la radiación electromagnética (HF y SHF), es sometida al impacto de fuertes campos magnéticos, que ejercen un efecto no contabilizado sobre el agua y sus impurezas.
4.24 Diferencias de energía entre los métodos de investigación acústica
Como resultado de la absorción de la señal por el medio que se investiga en los métodos de ultrasonido, la intensidad del sonido que se utiliza (I) constituye al menos 10−4–10−6 W/cm2. En comparación, señalamos que el motor de aviación crea un sonido de intensidad 10−4–10−6W/cm2 a una distancia de 5m. Bajo el efecto de una fuerte vibración, la estructura supermolecular de un líquido puede destruirse. Para los métodos de ultrasonido a la velocidad del sonido en el agua ? = 1498 m/s, la densidad de la energía del sonido (I/s) es igual a 7 × 10−9… 7 × 10−11 J/cm3. Esto constituye 1013-1011 kT / cm3 en T = 300 K (kT = 4.14 × 10−21 J).
Las investigaciones del agua a una frecuencia f = 1 kHz a la intensidad de señal 10-15-10-14 W/cm2, es decir, 10-20 dB (umbral de audibilidad de la señal de la frecuencia 1 kHz constituye 10-16 W / cm2[158]). La densidad de energía del sonido en este caso es igual a 6,6 × (10−19… 10−18) J/cm3, lo que constituye 2,4 × 104–2,4 × 106kT/ cm3.
La presión de la onda de sonido y la amplitud de la onda de sonido θ en las investigaciones de sistemas de agua a frecuencias de sonido es mucho menor que a frecuencias de ultrasonido. Por lo tanto, si a la frecuencia f = 1 mHz el valor θ = 1.7 × 104 Pa (= ρcω?, donde ρ - es la densidad del agua, ω = 2πf, ? es la amplitud del sesgo de las partículas), entonces a la frecuencia 1,000 Hz, la amplitud de la presión constituiría solo 5,5 × 10−4 Pa. Está claro que a presiones tan bajas uno debería esperar un efecto perturbador sustancialmente menor del método de investigación en estructuras débilmente unidas, como grupos de agua.
Un valor gráfico es la amplitud de deformación δ que se desarrolla en la onda de sonido (δ = ω?/s). Si a la frecuencia 1 mHz δ = 7.7 × 10−7 cm, entonces en la frecuencia de 1 kHz que usamos, el valor δ constituye solo 2.4 × 10−14 cm. En consecuencia, a señales de sonido de baja frecuencia y pequeña intensidad, la amplitud del sesgo y la deformación son sustancialmente más pequeñas que las dimensiones de los elementos estructurales del material que se investiga (es decir, átomos y moléculas de agua).
En el presente estudio se utilizó una unidad que produce una densidad extremadamente pequeña de energía sonora (del orden de 6,6 × 10−19… 10−18 J/ cm3). Por tanto, se introdujo una perturbación mínima en la estructura de la muestra de agua que se estaba investigando. Por ejemplo, para calentar el agua en 1 grado, es necesario gastar 0,18 J/cm3, que constituye el 0,42% de la energía de rotura de enlaces de hidrógeno. A la intensidad del radiador 10-15 W/cm2 (10 dB), la energía volumétrica del sonido (6,6 × 10-19 J/cm3) es insignificante en comparación con la energía necesaria para el calentamiento tangible del agua o la ruptura de los enlaces de hidrógeno. Debido a la amplitud infinitesimal, los desplazamientos por cizallamiento de las vibraciones sonoras, que determinan la viscosidad estructural, son difíciles de lograr. El coeficiente de absorción acústica α se calculó mediante la fórmula[159]
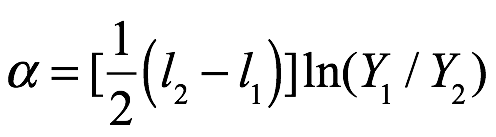 (3.7)
(3.7)
donde Y1, Y2 son valores de las señales para las distancias l1 y l2.
La figura 4.25a muestra la relación entre el valor de la señal Y y la distancia al radiador y el coeficiente de absorción acústica α, calculado utilizando estos datos a varias intensidades de señal del radiador en agua destilada a 20 °C e iluminado con una luz dispersa. de la superficie de la muestra ~ 50 T. La Tabla 4.13 contiene los resultados que determinan el valor α para el agua en diferentes tipos de tratamiento.
Los experimentos con agua han demostrado que el valor de las señales sonoras con intensidades menores y ausencia de efectos externos permanece constante durante un período de tiempo significativo. En condiciones experimentales idénticas (la potencia del radiador, la distancia al radiador, la temperatura, etc.) dependen de los cambios estructurales en el agua.
El valor Y disminuye al aumentar la distancia desde el radiador (ver Fig. 4.25a).
Debido a la atenuación del sonido, el coeficiente de absorción α disminuye al aumentar la intensidad de la señal del radiador. Se sabe que la absorción del sonido en líquidos está determinada principalmente por la viscosidad. Para agua con la frecuencia de 1 kHz, la absorción del sonido de relajación está ausente[160] y el valor α depende de la viscosidad estructural. Según la fórmula de Stokes, con un margen para la viscosidad volumétrica, un aumento de la absorción acústica es evidencia de un aumento de la viscosidad estructural:
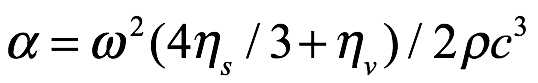 (3.8)
(3.8)
donde ηs - es la viscosidad de cizallamiento; ηV — es la viscosidad en volumen; ω = 2πf, ρ es densidad.
El concepto de viscosidad volumétrica como componente de la viscosidad estructural se utiliza en acústica para explicar las pérdidas de viscosidad bajo compresión integral, que acompañan a las ondas sonoras en un líquido. La energía de una onda de sonido se gasta en superar y aumentar la viscosidad del volumen ηV, como lo demostraron los experimentos, cuando la amplitud de vibración disminuye.
 21.25.44.png)
Fig. 4.25 Relación entre el valor de la señal Y (1, 2), el coeficiente de absorción α (3, 4) y la distancia entre el radiador y el receptor en agua destilada: (?) es la intensidad de la fuente de señal 1 × 10–15(1, 3), 2 × 10–15 w/cm2 (2, 4), f = 1 kHz, ? es 20 ° ? (el valor α se midió para una muestra iluminada); b) la muestra estuvo en la oscuridad o 18 h (1, 3), la muestra estuvo a la luz durante 4 h (2, 4), I — 2 × 10−15W/cm2, f = 1 kHz, ? = 20 ° ?.
La fórmula (4.25) se cumple a altas frecuencias e intensidades de sonido altas, pero no funciona en el caso de intensidades de sonido muy débiles. Si las medidas normales (a la intensidad de la señal 10−5-10−4 W/cm2 y la frecuencia 106-109 Hz) para el agua se cumple la relación ηs ≈ 3ηV, entonces a la intensidad del sonido 10−15 W/cm2 las fuerzas que provocan el movimiento longitudinal (a lo largo de la dirección de propagación de la onda) del líquido no son capaces de activar la transferencia de energía, debido a los efectos infinitesimales y la deformación de los enlaces moleculares en el agua. Un aumento de la viscosidad del volumen de cizallamiento a pequeñas intensidades de la señal puede ser determinado por un mismo mecanismo; por ejemplo, se puede observar la resistencia al cambio de orientación de las moléculas de agua o de los grupos de agua. Esto está relacionado con la rápida pérdida de intensidad de la señal observada experimentalmente (figura 4.25a) cuando se eliminan valores altos del coeficiente de absorción del radiador. Estos valores son comparables con el valor α para cristales líquidos y con α para frecuencias muy altas.
Se encontró que la absorción del sonido de baja intensidad en el agua depende de si la muestra de agua está en la oscuridad o está sujeta a la luz. La determinación de la absorción acústica se llevó a cabo después de que la muestra de agua estuviera en la oscuridad y después de 4 h de exposición a la luz (Fig. 4.25b). Para el agua destilada mantenida en la oscuridad, la curva de atenuación del sonido es menor que para la muestra iluminada. Esto es evidencia del hecho de que en el agua mantenida en la oscuridad (es decir, en condiciones en las que el agua está protegida de la influencia de las radiaciones electromagnéticas), la onda de sonido pierde más intensidad que cuando atraviesa el agua iluminada. Además, el coeficiente de absorción acústica calculado por la fórmula (4.12) para las muestras de agua oscurecida parecía mayor que para la muestra iluminada (ver Fig. 2.25b).
El papel del factor de iluminación/ofuscación en gran medida se manifiesta en mayores cantidades para agua más pura. Como puede verse en los datos de la Tabla 4.14, en el agua bidestilada la tasa de cambio del valor α es mayor que en el agua destilada, mientras que la tasa de cambio para el modo de iluminación no tiene efecto sobre la absorción acústica del agua del grifo. La Tabla 4.14 presenta los resultados de la medición de la absorción acústica por la frecuencia 1 kHz, intensidad 2 × 10−15 W/ cm2 a 20 °?. Un aumento de la intensidad de la señal del radiador (en los datos del experimento hubo un aumento de 15 veces) da como resultado una disminución del efecto de confusión y absorción del sonido por el agua (Tabla 4.14).
Tabla 4.14 Absorción del sonido por agua con diferentes tratamientos
 13.03.01.png)
Con base en los resultados obtenidos, se puede suponer que la macroestructura del agua, en condiciones de efectos externos mínimos (en el caso dado el agua está protegida de la radiación de la luz y el ruido de la radiación del sonido; una pantalla metálica también la protege de los efectos de la bajada). Radiaciones electromagnéticas de frecuencia), gradualmente (durante varias horas) se reconstruye, y las pérdidas de energía en el paso del sonido aumentan. Dado que la densidad media de la muestra, en términos de volumen, en la celda de medición no cambia, el efecto puede explicarse por un aumento de la viscosidad del líquido. Debido a la formación de pequeños grupos, la cantidad de moléculas de agua no unidas disminuye. Esto dificulta el aumento de la viscosidad y, de acuerdo con la Ec. (3.8), el paso de ondas sonoras.
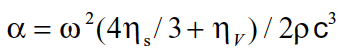
Para comprender el cambio del valor α en función del modo de iluminación, es necesario tener en cuenta ciertos factores. En primer lugar, se sabe que en términos de compresibilidad de pequeños efectos mecánicos, los líquidos se aproximan a los cuerpos sólidos. Esto está determinado por el hecho de que no hay suficiente tiempo para la redistribución de moléculas bajo influencia externa. Las moléculas, como en los cuerpos sólidos en pequeñas amplitudes de variaciones, casi no cambian de posición. La energía del sonido, en este caso, se absorbe al máximo (3.8) debido a la fuerza predominante de fricción interna (es decir, viscosidad acústica volumétrica ηV) cuya contribución aumenta con respecto a la fuerza de la onda sonora a medida que disminuye la intensidad del sonido. Por tanto, los valores α parecen ser altos y aumentan en ausencia de iluminación debido a la formación de estructuras de racimo que aumentan la viscosidad del agua.
Habiendo comparado los resultados dados en las Figs. 4.25a, b, se puede concluir que incluso el sonido de poca potencia ejerce alguna influencia sobre la macroestructura del agua, aunque esta influencia no es tan fuerte como la de la radiación luminosa. De los datos de la Tabla 4.14 se deduce que, en primer lugar, el agua del grifo tiene un coeficiente de absorción de sonidos débiles más alto que el agua destilada. En segundo lugar, los efectos del factor de confusión/exposición a la luz en el agua del grifo son mucho más débiles. Ambos efectos, quizás, están relacionados con el hecho de que en el agua del grifo una mayor concentración de impurezas de iones metálicos y moléculas de agua en mayor grado están unidas a hidratar capas de iones que a grupos, ya que la energía de hidratación de iones es sustancialmente mayor la energía de los enlaces H del agua. El impacto de los iones es indicativo de un mayor coeficiente de absorción de señales débiles por el agua bidestilada, en comparación con la destilada (para muestras no sujetas a exposición). Por tanto, se puede concluir que si el agua no fuera sometida al efecto activador de la luz, la formación de racimos (o estructuración general si se toman en consideración otros modelos) procedería con más fuerza en ausencia de iones, que reconstruyen la estructura del agua en el estructura de conchas de hidratos.
La destrucción parcial de pequeños racimos durante la transición del agua de un ambiente oscuro a uno claro hace que se forme una estructura suelta; es decir, cuando se expone a la luz, el agua adapta una estructura característica de las temperaturas más elevadas. Las nociones sobre la influencia de los iones en la estructura y la temperatura del agua[161] pueden llevar a inferir que en condiciones de exposición a la luz la temperatura del agua es más baja que en la oscuridad. Esto significa que, por ejemplo, a 20 °C en la oscuridad, el agua posee una estructura que se correlaciona con una temperatura más baja. Ciertamente, el efecto de variación de la temperatura del agua "estructural" debido a la formación de grupos de agua es sustancialmente menor que debido a la formación de capas de hidratos de iones debido a la energía del efecto de polarización de los iones, a juzgar por los efectos del calor en las soluciones, es mucho más grande que el de los clústeres.
Dada la oscuridad simultánea del agua y su magnetización durante 16 h por el campo de inducción ~ 80 mT, se puede observar un aumento en la absorción de una señal de sonido (ver Tabla 4.14). Después de la eliminación del campo magnético y la exposición a la luz, la señal se reduce al valor característico del agua destilada en condiciones de iluminación. En consecuencia, en el caso de un efecto prolongado, el campo magnético permanente conduce a un aumento de la viscosidad del agua de un entorno oscuro. Quizás en el campo magnético se promueve la formación de cúmulos procediendo lentamente a la polarización de la migración[162].
La polarización implica grupos cargados, es decir, partículas moleculares dipolo que llevan cargas eléctricas como resultado de su compensación estequiométrica incompleta durante la formación de un grupo. La compensación incompleta de las cargas de las moléculas de agua se produce como resultado de la deformación de las moléculas o debido a la desigualdad del estado de las moléculas en el interior y en la superficie de un grupo de agua. La formación de la carga superficial de un cúmulo provoca su polarización bajo el efecto de campos externos.
Un grupo puede modelarse como una partícula dispersa con una doble capa eléctrica que asegura las fuerzas de repulsión de los grupos vecinos. En este caso, el agua puede considerarse como una suspensión de grupos y la concentración de la fase dispersa (es decir, grupos) en dicha suspensión depende de las condiciones externas (temperatura, presencia de impurezas, etc.). El campo eléctrico o magnético externo polariza agrupaciones similares a las partículas de la fase dispersa. Creemos que estas posibilidades del modelo de clúster son ventajosas. Lo más probable es que el modelo continuo del agua estructurada sea más adecuado para bajas temperaturas; se aplica mejor, en particular, a condiciones de sobreenfriamiento del agua[163].
Como resultado de los experimentos, se deduce que incluso un campo magnético permanente débil bajo un efecto prolongado es capaz de afectar la estructura del agua destilada, que no está sujeta al impacto de las radiaciones. Como han demostrado los experimentos, el tratamiento de tres días con el campo magnético no dio como resultado el cambio del valor α. Los cambios de estructura en la muestra de agua oscurecida, que habían tenido lugar bajo los efectos del campo magnético, fueron desapareciendo gradualmente y la estructura del agua volvió a su estado inicial. Un efecto de corta duración en el agua por tratamiento de ultrasonido (f = 22 kHz, I = 0,1 W/cm2, 0,5 min) aceleró el proceso de disminución del valor α al valor característico de la muestra de agua iluminada. Esto también corrobora el hecho de que bajo los efectos de la luz la viscosidad de la muestra disminuye, debido a la destrucción de la estructura del racimo. El tratamiento con ultrasonido acelera el proceso de destrucción de estructuras secundarias.
Así, gracias a un efecto perturbador muy débil del método de investigación dado sobre el agua, se registraron los valores de α, atestiguando los cambios de viscosidad determinados por la reestructuración de la estructura del racimo bajo el impacto de diferentes iluminaciones de la muestra.
Considerando los resultados obtenidos se puede concluir que el estado principal de la estructura del agua debe considerarse protegido de la luz, vibraciones y diversas radiaciones electromagnéticas. Este estado también implica la ausencia de sustancias disueltas. En este caso, se cumple un verdadero equilibrio entre la estructura del grupo de agua y las moléculas de agua no unidas. La iluminación u otros efectos dan como resultado una destrucción gradual de los racimos y la transición del agua del estado principal a otros estados característicos del efecto dado.
4.25 La investigación teórica de las interacciones de los grupos de agua con el ozono
Las moléculas de ozono, como se sabe, juegan un papel importante en la atmósfera terrestre. Muchos autores estudiaron la interacción del ozono con los compuestos atmosféricos, quienes señalaron que los complejos de ozono con el agua son formaciones clave en las reacciones de la capa de ozono[164]. Bajo el efecto de la radiación ultravioleta (UV), el ozono atmosférico entra en reacción con varias moléculas y radicales de la atmósfera. Se sabe que la radiación UV solar con una longitud de onda de 185.9 nm provoca la formación de ozono a partir del oxígeno y con una longitud de onda de 200–320 nm, su descomposición. El radical aniónico ozonido es un producto primario de la descomposición del ozono
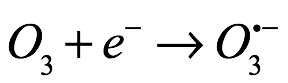
La siguiente formación y otras partículas de radicales libres son posibles con la participación de ozono
 14.39.37.png) etc[165].
etc[165].
Esto se puede ilustrar usando oxígeno singlete como ejemplo. Este oxígeno se forma como resultado de la fotólisis UV del ozono. El oxígeno singlete, a su vez, reacciona con las moléculas de agua con la formación del radical hidroxilo oxidante principal. Las reacciones dadas están representadas por el siguiente esquema:
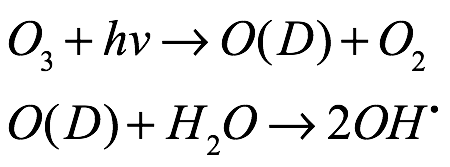
Cabe señalar que el ozono puede coexistir con oxígeno singlete y molecular cuya relación en una carga de gas es igual a
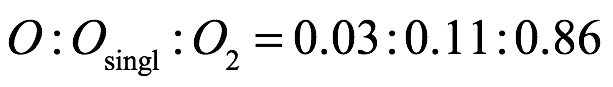
En este caso, el ozono medio en el agua no puede considerarse independiente del pH porque los hechos experimentales confirman el impacto del pH sobre la actividad preferencial del ozono por mecanismo radical o heterolítico. También es posible la formación de radicales hidroxilo a partir de complejos solvatos de ozono:
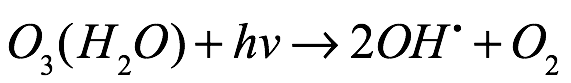
A este respecto, la información sobre la naturaleza de la interacción de los complejos de ozono con el agua es especialmente importante, ya que la capa de hidrato puede desplazar sustancialmente el pico de absorción y por tanto afectar a la formación de radicales libres.
El estudio de la interacción del ozono con el agua, propuso un modelo en el que ambos átomos de oxígeno terminales están orientados hacia el lado de uno de los átomos de hidrógeno del agua[166]. Otro estudio propuso un modelo en el que el hidrógeno del agua forma un enlace de hidrógeno con solo uno de los átomos de oxígeno terminales de la molécula de ozono[167]. Posteriormente, se propuso un modelo “dipolar” del complejo de agua con ozono, en el que una molécula de agua junto con el átomo central de ozono forman el plano de simetría Cs[168].
Por tanto, la información sobre las estructuras de los complejos de O3 (H2O) no proporciona una idea única sobre la naturaleza de la interacción de las moléculas de ozono con el agua. Las estructuras de los grupos de agua con ozono se optimizaron completamente al nivel teórico de la teoría de perturbaciones de segundo orden de Møller-Plesset. El conjunto básico de funciones de onda 6–311 ++ G(d, p) de tipo gaussiano, con la inclusión de funciones de difusión y polarización, se eligió tanto para la optimización como para la determinación de frecuencias. Las frecuencias de vibración se calcularon basándose en estructuras optimizadas mediante el método mencionado anteriormente. La representación gráfica de los grupos de agua se obtuvo mediante el software Molden[169]. La energía de formación del complejo agua-ozono, o la energía de enlace ES, se calculó sobre la base de la energía del estado de equilibrio del grupo y el estado principal de la molécula de ozono mediante el método MP4 (SDQ) con un conjunto de bases mejorado
6–311 + + G (2d, 2p):
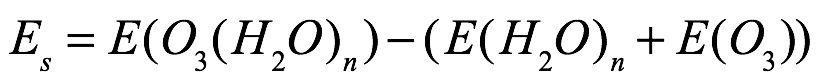 (3.9)
(3.9)
El parámetro del criterio de convergencia para la energía y el gradiente del campo autoconsistente se establecieron en 10-7. Hemos optimizado varios de los modelos O3-H2O (figuras 4.26–4.29). Para confirmar el mínimo global, se realizó el análisis de frecuencia de las estructuras de agua con ozono, obtenido en optimización. Como resultado, se determinaron dos complejos estables (figura 2.26). El primer modelo (figura 4.26a) representa una estructura en la que H2O y O3 se encuentran virtualmente en un plano, y un átomo de hidrógeno se gira hacia el lado del átomo de oxígeno terminal del ozono. Por tanto, el enlace de hidrógeno aparece entre un átomo de hidrógeno del agua y el átomo de oxígeno terminal en el ozono. Sin embargo, la longitud de este enlace, 0.227 nm, excede sustancialmente la longitud del enlace de hidrógeno en los grupos de agua[170], y debido a esto, la energía de su formación (9.15 kJ/mol) es inferior a la energía del enlace de hidrógeno en conglomerados de agua.
El segundo modelo (figura 4.26b) posee una gran cantidad de energía de enlace (9.78 kJ/mol) y tiene simetría ?s. El eje de simetría pasa por el átomo de oxígeno central del ozono y el átomo de oxígeno del agua. Según los resultados de los cálculos, dicha configuración eclipsada, en la que los átomos de hidrógeno son opuestos a los átomos terminales del ozono, es la forma más estable de complejos de agua con ozono en la proporción 1:1.
La forma trans indicada en la bibliografía (figura 4.26a) también se reprodujo en el estudio actual[171]. Sin embargo, como han demostrado los cálculos, la configuración dada es metaestable, lo que se indica por la presencia de la frecuencia imaginaria en el análisis vibracional. La energía del enlace de tal complejo constituye solo 8.02 kJ/mol. Desde nuestra perspectiva, la orientación del dipolo (figura 4.26b) es una estructura inestable y simplemente un estado de transición a la configuración eclipsada.
El análisis de frecuencias armónicas de la estructura dipolo reveló varias frecuencias con un valor negativo. A la menor perturbación (por ejemplo, al eliminar la simetría) el complejo dado adquiere la configuración de la forma eclipsada.
 17.58.04.png)
Fig. 4.26 Complejo de ozono con el monómero de agua O3 (H2O) (a) y configuración protegida del isómero O3(H2O) (b).
 17.58.10.png)
Fig. 4.27 Complejo de ozono con el dímero de agua O3 (H2O)2 (a), con el trímero de agua O3 (H2O)3 (b), con el tetrámero de agua O3 (H2O)4 (c) y el isómero O3 (H2O) 4 (d).
 17.58.17.png)
Fig. 4.28 Configuración "Trans" (a) O3 (H2O) y orientación "dipolo" (b) isómero O3(H2O)
 17.58.24.png)
Fig. 4.29 Complejo de ozono con el pentámero de agua O3 (H2O)5 (a) y con la forma cúbica del hexámero O3(H2O)6 (b).
Las constantes de rotación para dos configuraciones estables se dan en la Tabla 4.15. Las constantes de rotación obtenidas para la configuración eclipsada concuerdan directamente con los datos experimentales. La estructura optimizada del complejo O3 (H2O)2 se muestra en la Fig. 4.30a. Consiste en el dímero de agua y el ozono en los que el átomo central de oxígeno se coordina con el átomo de oxígeno del monómero de agua (el donante del enlace de hidrógeno), donde ro-o = 0.284 nm. Al mismo tiempo, el átomo de hidrógeno, el monómero aceptor, forma un enlace de hidrógeno débil con uno de los átomos terminales de oxígeno del ozono.
La longitud del enlace de hidrógeno en el dímero de agua de un complejo con ozono constituye 0,194 nm, que es comparable con la longitud del enlace de hidrógeno en el dímero libre de 0,195 nm. Cabe señalar que el dímero de agua se une más fuertemente al ozono que el monómero. Por tanto, la energía de disociación del O3(H2O)2 constituye 15,49 kJ / mol.
Los complejos agua-ozono optimizados con el número de monómeros de agua 3–6 son compuestos en los que las moléculas de agua tienen una forma cíclica del trímero, pentámero, isómeros de tetrámero y hexámero estudiados anteriormente. En todos los complejos enumerados, una molécula de ozono está coordinada a través del enlace oxígeno-oxígeno por solo un monómero de la composición de la estructura del agua ( figuras 4.27, 4.28, 4.29).
La distancia intermolecular más cercana (0,283 nm) entre la molécula de agua de unión y el ozono en los complejos O3 (H2O) n (n = 1… 6) se obtuvo para O3 (H2O)2. La distribución de la carga en los átomos de oxígeno del ozono y el monómero de agua que interactúa con él se da en la tabla 4.15. Como puede verse, la carga del átomo central de oxígeno en el ozono libre constituye + 0.321, mientras que en los complejos con racimos de agua aumenta apreciablemente. Según los datos obtenidos, el mayor valor positivo para el átomo central de oxígeno del ozono se logra en complejos con el dímero y tetrámero de agua. La distribución de la carga obtenida es el resultado de la interacción de Coulomb con la molécula de unión del monómero del grupo de agua.
Así, como resultado de los cálculos teóricos ab initio en el estudio de los complejos de ozono con los grupos de agua ?3 (H2O) n para n = 1… 6 se obtuvieron los siguientes resultados. En la relación ozono: agua = 1: 1 se descubrieron dos configuraciones obsoletas con la energía de enlace: 9,15 y - 9,78 kJ/mol (Tabla 4.15). Es de destacar que la estructura, en la que el ozono se une a la molécula de agua como el enlace de hidrógeno, posee un valor energético total más alto. La energía más alta del enlace n de O3 (H2O) (- 90,94 kJ/mol) se obtuvo en el compuesto con el isómero de hexámero, y la más pequeña (-9,15 kJ/mol) se obtuvo en el complejo con el monómero. A excepción del dímero de agua en todos los grupos de agua estudiados por nosotros, el ozono está unido a uno de los monómeros más cercanos exclusivamente a través del enlace O–O, que fue testificado a partir de datos estructurales así como por la distribución de carga en los átomos.
 17.58.30.png)
Fig. 4.30 Isómero O3 (H2O)6
Tabla 4.15 Constantes de rotación para el complejo O3(H2O)
 13.09.54.png)
 17.58.54.png)
Así, la interacción O–O electrostática prevalece sobre la formación de un enlace de hidrógeno en los complejos de ozono con los grupos de agua. El presente estudio corrobora la hipótesis, expuesta anteriormente, sobre la transformación del ozono que involucra aniones hidroxilo, cuya etapa inicial puede escribirse como
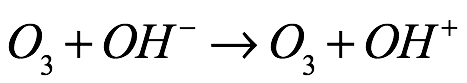
Como ya se ha señalado, en la complejación del ozono con agua se incrementa una carga positiva parcial en el átomo central de oxígeno del ozono, lo que de una manera sustancial conduce a la interacción con el anión hidroxilo cargado negativamente. Al mismo tiempo, la presencia de la carga fraccional de Coulomb en el átomo central de oxígeno del ozono inhibe la formación del enlace de hidrógeno. De este hecho se deduce que la solubilidad relativa del ozono en el agua, en primer lugar, está determinada por la interacción dipolo-dipolo de Oc—Ow más que por la presencia de enlaces de hidrógeno, que, según los cálculos, tienen una comparativamente pequeña energía de formación.
4.26 Impacto de la temperatura en los racimos de agua
El uso reciente de métodos de investigación ópticos asistidos por computadora ha hecho posible estudiar la influencia de la temperatura en la estructura del grupo de agua. Para ello se ha utilizado un microscopio con láser (λ = 633 nm). Un rayo láser después de pasar por un sistema de lentes con un aumento total de 20 veces viajó a través de una bandeja espectrométrica llena con una muestra y luego se proyectó en la pantalla.
El termostato se usa para ajustar la temperatura dentro del rango de 10 a 45 °C y se mantiene con una precisión de ± 0.2 °C. La pequeña profundidad de enfoque (~1 μ) de la lente del microscopio hizo posible observar elementos de contraste en la sección elegida de la solución. La imagen que se proyecta en la pantalla es el resultado de la dispersión de la luz del rayo láser desde los límites de fase de los elementos líquidos de dimensiones microscópicas. Estos elementos pueden ser partículas de polvo, burbujas de aire y otros asociados de moléculas de agua, que son diferentes del agua no asociada en la interfaz por el coeficiente de refracción de la luz.
La imagen de la pantalla fue leída por una cámara de video e ingresada a la computadora. Luego se convirtió en blanco y negro y se transformó en una matriz de un sistema bidimensional y se analizó mediante software de computadora. El resultado del análisis se obtuvo en forma de la concentración total de conglomerados y la concentración de conglomerados de determinadas dimensiones (4 a 40 μ). El aumento del microscopio limitó el tamaño inferior de los objetos observados al tamaño de 2 μm, lo que permitió descartar el recuento por el software de microimpurezas como microburbujas de gas, partículas coloides, etc. El microscopio se calibró mediante un micrómetro de fotomicrografía OMP-2. Luego, la escala se proyectó en una pantalla y luego se envió mediante una cámara de video a una PC. Finalmente, la calibración de los elementos de la imagen en la pantalla se expresó en píxeles (en el caso dado, 1 píxel = 2 μ).
Fig. 4.31 Dependencia de la temperatura de la concentración total de GHWC: 1 — agua destilada, 2 — agua magnetizada, 3 — jugo natural de abedul.
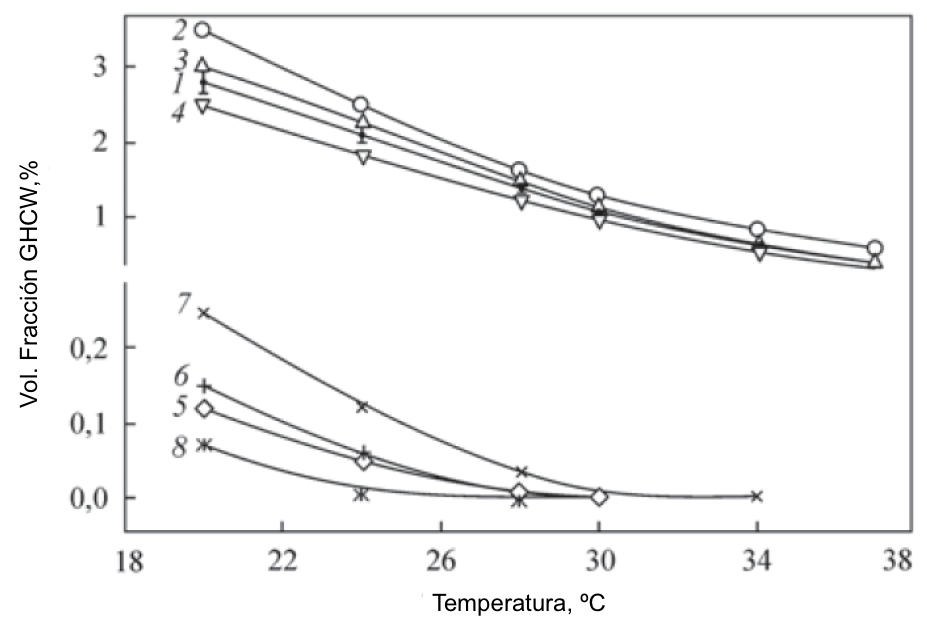
Fig. 4.32 Dependencia de la temperatura del contenido de GHWC de tamaño 4 (1-4) y 36 μ (5-8) en las soluciones LiCl (1, 5), NaCl (2, 6), KCl (3, 7) y RbCl (4, 8)
La figura 4.31 muestra las curvas típicas de la concentración total de conglomerados (es decir, el contenido total de GHWC de dimensiones 2 40 μ) a partir de la temperatura dentro del intervalo 15–38 °C. Se obtuvo una muestra de agua destilada magnetizada al pasar el agua por una columna de 2.5 cm de diámetro y 10 cm de alto, y luego se llenó la columna con bolas de 3 mm de diámetro hechas de material magnético de hierro-níquel, creando inducción magnética en un espacio entre partículas de hasta 100 mT. La magnetización se llevó a cabo en condiciones estáticas durante un cierto período de tiempo. Se magnetizó agua destilada en una columna (en el caso dado durante 27 min) después de que se tomó la sonda y se determinó la GHWC. El líquido poroso de un árbol se probó como un líquido vegetal natural que no contiene GHWC. Para este propósito, se tomó la savia de un abedul. Antes de medir el GHWC, esta savia se filtró a través de un filtro cerámico poroso fino obtenido por sinterización de partículas minerales con tamaños de 10 a 40 μ. Como se puede ver en la Fig. 3.20, la dependencia de la temperatura del contenido de GHWC en el jugo de abedul natural por forma es similar a la curva del agua. Así, se puede llegar a la conclusión de que, en primer lugar, el GHWC está contenido en el líquido poroso de las plantas; en segundo lugar, la cantidad de GHWC en el líquido de las plantas depende de la temperatura. La figura 4.31 muestra las dependencias de la temperatura para grupos de tamaño 4 y 36 μ en las soluciones 0.01 M de LiCl, NaCl, KCl y RbCl. El error característico del experimento, estimado en el caso dado en un 5%, se indica en la curva 1. La concentración se eligió en base a la afinidad de la solución 0,01 M de NaCl por el organismo humano; por lo tanto, fue necesario evaluar el contenido de GHWC en dicha solución a diferentes valores de temperatura. Varios cloruros investigados corresponden a una disminución de la fuerza de polarización del catión, que está determinada por un aumento de su radio y aparece en muchas propiedades de las soluciones acuosas[172]. Como puede verse en las Figs. 4.31 y 4.32, para todos los sistemas de agua investigados, el contenido de GHWC a un aumento de la temperatura disminuye. El contenido total de racimos al calentarlo puede disminuir de 40 a 15 º? a 2–2,5% a 38 ° C.
El contenido de GHWC de varios tamaños a un aumento de la temperatura disminuye de manera no uniforme. Como se puede ver en la figura 4.32, un calentamiento más fuerte afecta a los grupos grandes. En realidad, a la temperatura de 30 °C y superiores, los grupos de dimensiones de 36 μ ya no existen. Al mismo tiempo, se conserva una cierta cantidad de racimos de tamaño 4 μ, a pesar de calentar la solución a 38 °C. Cabe señalar que a la temperatura del organismo humano de 36 a 39 °C en soluciones acuosas, los grupos del tipo GHWC están ausentes. A una temperatura más baja (30–32 ° C) en las soluciones se conservan GHWC de dimensiones más pequeñas. Una disminución del contenido de GHWC con un aumento de la temperatura indica la destrucción de los grupos bajo el impacto de la energía kT. El contenido de partículas en la solución, en el caso actual denominado clusters, depende de la energía de la interacción de moléculas de agua como GHWC, capaces de formar clusters. La condición para la formación de racimos es un exceso de moléculas de agua de un cierto nivel de energía (barrera de energía), lo que resulta en su interacción con la formación de GHWC. Cuanto mayor sea la temperatura de la solución, mayor será la energía de la interacción de las moléculas para alcanzar y superar la barrera de formación de grupos.
A una disminución de la temperatura de la solución, la energía de desorden térmico (kT) disminuye; por lo tanto, es más fácil para las moléculas de agua superar la barrera energética y se forman GHWC mayores. Por tanto, la formación de GHWC depende de la temperatura de interacción molecular, que, a su vez, favorece el cambio del contenido de racimos en la solución. Al investigar GHWC, se puede observar un resultado final de dicha interacción, a saber, el cambio de concentración de los conglomerados. El valor de la concentración total de racimos muestra cuánta agua participa en la formación de GHWC con respecto a la cantidad de agua que no interviene en este proceso.
Para determinar la energía de activación de destrucción de conglomerados, se construyeron las dependencias de temperatura en las coordenadas de Arrhenius[173] (Figuras 4.33 y 4.34). La curva 1 (Fig. 3.34) muestra el error de determinación que constituye el 5%. Como puede verse a partir de los datos dados en las Figs. 4.33 y 4.34 todos los puntos de los sistemas investigados se colocan en líneas rectas, que son individuales para cada sistema. Esto corrobora la aplicabilidad de la ecuación de Arrhenius a los sistemas hídricos dados y muestra que cada uno de ellos se caracteriza por cierta energía de activación de racimos destructores del tipo GHWC. Según la ecuación de Arrhenius,
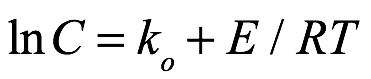 (3.10)
(3.10)
donde C - es la concentración de GHWC, mol/dm3; k0: es el coeficiente que caracteriza la frecuencia de colisión de moléculas en reacción; ? - es la energía de activación, kJ/ mol; R — es la constante universal de los gases, J/(mol · ?); T — es la temperatura, ?. Para calcular la energía de activación emplearemos la relación gráfica ln C — 1/T y las siguientes relaciones: tg α = ΔY/ΔX; E = R tg α (R = 8,31 J/(mol · ?)). Los resultados de la evaluación de la energía de activación de destrucción de conglomerados se dan en la Tabla 4.16. De los datos de la tabla 4.16 se deduce que el agua pura posee la mayor resistencia al calentamiento (es decir, la mayor energía de activación para destruir GHWC). En las soluciones de las sales NaCl y KCl la energía de activación de los racimos es menor que en el agua destilada. Sales como LiCl y RbCl son las que más conducen a la destrucción de racimos del tipo GHWC. Cabe señalar que en la destrucción de GHWC se revela la capacidad conocida de los cationes de carga única para la hidratación positiva o negativa. El catión de litio es el más hidratante y destruye los racimos reteniendo moléculas de agua en la capa de hidrato. Al contrario, el Rb +, que se caracteriza por una hidratación negativa, destruye los racimos, probablemente debido a una fuerte disminución de la densidad media de la disposición de las moléculas de agua cerca del catión.
 18.36.47.png)
Fig. 4.33 Dependencia de la temperatura de la concentración total de GHWC en las coordenadas de Arrhenius: 1 — agua destilada, 2 — agua destilada magnetizada, 3— jugo de abedul natural.
 18.36.51.png)
Fig. 4.34 Dependencia de la temperatura del contenido total de GHWC en soluciones de cloruros de metales alcalinos (concentración 0.01 mol/dm3) en las coordenadas de Arrhenius: 1 — agua destilada, 2 - LiCl, 3 — NaCl, 4 — KCl, 5 — RbCl
La magnetización del agua disminuye la energía destructiva de activación de GHWC al nivel del impacto del aditivo NaCl. Así, al añadir sales al agua o magnetizar el agua potable el contenido de GHWC en la misma disminuye (en primer lugar de grandes dimensiones), lo que es similar al tratamiento del agua por calentamiento a> 30 °C. Para lograr un alto grado de descomposición del agua de los grupos del tipo GHWC, el agua debe calentarse a 36 °C y más. Por lo tanto, en un organismo humano, el agua se encuentra en una forma desclusterizada en términos de GHWC. A diferencia de esto, el agua en los canales de las plantas puede contener GHWC en concentraciones sustanciales. En conclusión, podemos señalar la capacidad del agua para la relajación del contenido de GHWC. Para ello, se realizaron los siguientes experimentos con el agua de las salas de bombas cuya mineralización total constituía 290 mg/dm3 ( Tabla 3.16). El agua se calentó a ebullición y luego se enfrió a 20 ° C a diferentes velocidades (experimentos 1 y 2). En el otro caso, las muestras de agua se enfriaron a 0 °C, luego se congelaron y se calentaron a varias velocidades a 20 °C (experimentos 3 y 4). Después de alcanzar la temperatura de 20 °C en cada una de las cuatro muestras, determinamos la cantidad de GHWC durante ciertos períodos de tiempo (0, 10 y 20 min). Como han mostrado los resultados de los experimentos (Tabla 4.17), en las muestras de agua obtenidas por su calentamiento de 0 a 20 ° C el contenido total de racimos del tipo GHWC es mayor que en las muestras obtenidas por enfriamiento de agua de 100 a 20 °C. Esto concuerda con los datos obtenidos anteriormente en términos de dependencia de la temperatura del contenido de los clústeres, que muestra que a un aumento de temperatura los GHWC se destruyen y su concentración cae a cero. A una tasa más alta de enfriamiento por agua (experimento 2), el contenido de racimos es ligeramente menor que en enfriamiento lento (experimento 1). En consecuencia, el enfriamiento lento del agua desagrupada favorece los mecanismos de restauración de las estructuras de los grupos en el agua. La capacidad del agua para mantener la estructura anterior en la transición de la muestra de la temperatura baja a la habitación uno se manifiesta aún más conspicuamente (experimentos 3 y 4). Si el agua enfriada se calienta rápidamente (experimento 4) (se refiere en su totalidad al “agua de fusión”), se inhibirá la destrucción de GHWC. La concentración de GHWC durante algún tiempo permanece más alta en comparación con la concentración característica de la concentración dada. En consecuencia, los procesos de formación y destrucción de cúmulos, tipo GHWC, toman bastante tiempo (del orden de segundos y minutos), lo que puede explicarse por la orientación y polarización migratoria de las partículas y la presencia de fenómenos de relajación característicos del agua[174]. La capacidad del agua para disminuir la cantidad y tamaño de GHWC al calentarse, en nuestra opinión, es una de sus nuevas anomalías estructurales.
Cabe señalar también que las variaciones estudiadas de la estructura del agua expresadas en un contenido disminuido de GHWC de diversas dimensiones durante el calentamiento a la temperatura de 32–38 °C probablemente se encuentran detrás de una serie de otras anomalías del agua ya conocidas. Se ha encontraron que dentro del intervalo de temperatura 35–40 °C el agua se caracteriza por el mínimo de capacidad calorífica específica[175]; el agua caliente (máximamente desagrupada) puede congelarse más rápido que la fría; la viscosidad del agua a> 32 °C depende poco de la presión, mientras que dentro del intervalo de temperaturas de 30–50 °C, la compresibilidad del agua es mínima. A las dimensiones de temperatura> 38 °C, la velocidad del sonido en el agua disminuye drásticamente, lo que puede deberse a un contenido disminuido de GHWC de grandes dimensiones.Esto, a su vez, permite inferir que los GHWC están involucrados en la transferencia de ondas sonoras en mayor grado que el agua agotada del racimo.
Tabla 4.16 Impacto de las sales (concentración 0.01M) o magnificación en la activación de energía de destrucción de conglomerados de agua
 13.14.26.png)
Tabla 4.17 Contenido total de GHWC en el agua de diferentes modos de tratamiento
 13.14.37.png)
4.27 Estructura de conglomerados de agua habitual, pesada y ligera
En la corriente principal de investigación[176] para el estudio de conglomerados en varios sistemas de agua, los autores consideraron el impacto de cambiar la temperatura para el contenido de conglomerados en agua pesada. (99.99% de D2O) y en muestras de agua con diferente contenido de deuterio. Se han investigado muestras de agua ligera, pesada y habitual. El agua ligera contenía 54 ppm o 0.0053997% de deuterio, la pesada: el 99.99% de D2O bidestilada habitual (agua de la sala de bombas): 154 ppm, que corresponde al 0,015398% de D2O. Se diluyó agua pesada con destilada en las proporciones que se dan en la Tabla 4.18. En este caso se obtuvo una muestra con cierto contenido de deuterio, que se colocó en una placa de laboratorio de espectrometría de medición y se realizaron experimentos según la técnica descrita en por Goncharuk[177].
Al estudiar la dependencia de la temperatura del contenido de los racimos, tipo GHWC, en el 99.99% del agua pesada, se cumple el modo de su calentamiento o enfriamiento. Se utilizó un termostato para ajustar la temperatura dentro del intervalo de 15 a 37 ± 0.2 °C (modo de calefacción) y de 37 a 10 ± 0,2 °C (modo de enfriamiento). Los difractogramas se midieron cada tres grados (por separado en el modo de calefacción y en el modo de enfriamiento). La muestra se mantuvo a la misma temperatura durante al menos 10 min.
Tabla 4.18 Preparación de la solución para el experimento
 13.19.12.png)
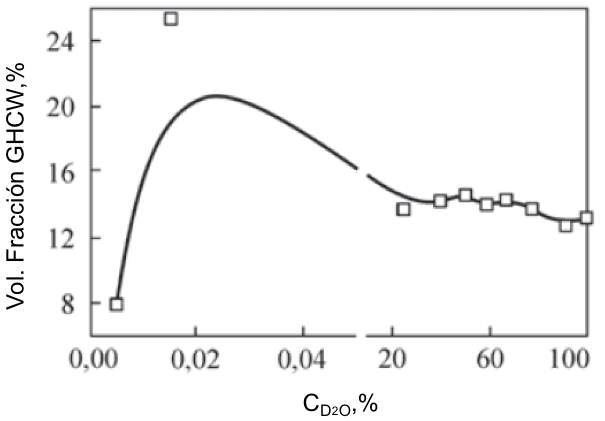
Fig. 4.35 Relación entre el contenido de GHWC en el agua y la concentración de D2O en la muestra.
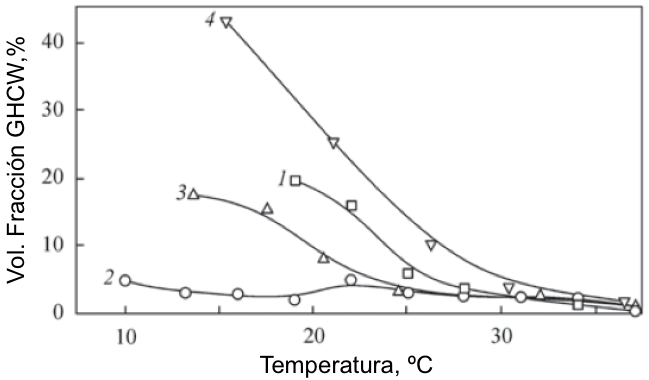
Fig. 4.36 Dependencia de la temperatura del contenido de los grupos de tipo GHWC en varias aguas: 1 — agua pesada, fracción total de D2O 99.99% (2 × 106 ppm D2O), 2 — bidestilada (154 ppm D2O), 3 — ligera (54 ppm D2O), 4: destilado (154 ppm D2O).
La Figura 4.35 muestra la relación entre el contenido de GHWC en el agua y el contenido de D2O para una amplia región de concentración de deuterio. La figura 4.35 muestra la dependencia de la temperatura del contenido de racimos en muestras de agua con diferentes contenidos de deuterio; Fig. 4.36: el impacto del calentamiento y el enfriamiento en el GHWC en el 99,99% de D2O. En la figura 4.37 se muestran las curvas de diseño para el contenido en el agua de grupos del tipo GHWC dependiendo de la viscosidad del agua en función de la temperatura.
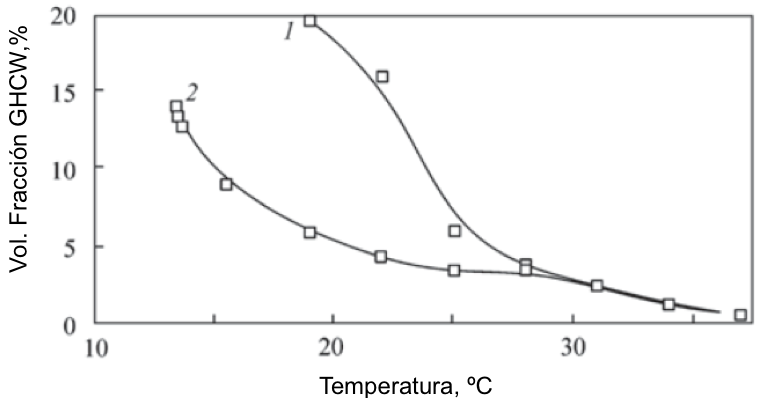
Fig. 4.36 Dependencia de la temperatura del contenido de racimos en agua pesada (99.99% D2O): 1 — calentamiento, 2 — enfriamiento.
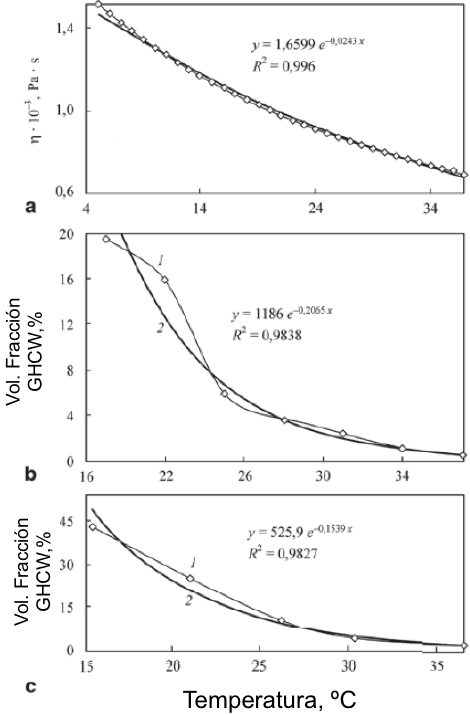
Fig. 4.37 Curvas aproximadas de la relación entre la viscosidad del agua y la temperatura (?), datos experimentales de calentamiento de agua pesada (b) y destilada (c): 1 — datos experimentales, 2 — línea de tendencia.
Dado que los enlaces de deuterio en agua pesada son más fuertes que los enlaces de hidrógeno normales[178], es de esperar que en agua pesada la concentración de GHWC o sus dimensiones sean mayores. Los experimentos mostraron que no se observan tales efectos. Se sabe que el 99% del deuterio en el agua habitual está representado en forma de DHO. La existencia simultánea de enlaces de deuterio e hidrógeno en las soluciones de DHO, en nuestra opinión, conduce a la formación de grupos debido a un mayor efecto de polarización de los dipolos mixtos.
Como puede verse en las relaciones gráficas dadas, la adición de agua habitual a D2O no cambia efectivamente el contenido de racimos gigantes en la región de volumétrico de 99.99 a 25%. D2O. Se refiere a fracciones de GHWC de 4 a 40 μ. La figura 3.35 muestra la dependencia de la temperatura del contenido de los conglomerados. Puede verse en la Fig. 4.35 que en agua destilada se puede observar la cantidad máxima de racimos; en agua bidestilada, por el contrario, la cantidad de racimos está en un nivel muy bajo y efectivamente no depende de la temperatura. Estos datos se pueden explicar por el hecho de que se utilizó agua bidestilada recién preparada, es decir, se preparó con un tratamiento térmico preliminar. El documento muestra que, como resultado del tratamiento térmico, los GHWC se destruyen. Para la restauración de la cantidad inicial de racimos, tipo GHWC, tal agua necesita cierto tiempo para asentarse en un estado de calma.
Esto es necesario para los lentos procesos de orientación y polarización de la migración, que conducen a la autoorganización de las moléculas de agua en grupos. A partir de datos experimentales sobre el impacto del calentamiento y enfriamiento del agua pesada (Fig. 4.36), se puede ver que la participación volumétrica total de GHWC debido al calentamiento disminuye del 20 al 0,5%. Si se enfría por agua de 37 a 28 °C, puede resultar obvio que efectivamente no se puede observar ningún cambio, es decir, la destrucción ocurrida de los racimos y el cambio de temperatura dentro de este intervalo no afectan la restauración de su cantidad inicial. En enfriamiento, la curva 2 (Fig. 4.36) no coincide con la curva 1, lo cual es una evidencia de la formación de conglomerados en una cantidad mucho menor. La presencia del bucle de histéresis apunta al hecho de que la formación de grupos sigue el otro mecanismo en comparación con el proceso de su destrucción (curva 1).
Lo más probable es que ambas curvas converjan en una a temperaturas más bajas. Las curvas para agua destilada (154 ppm) coinciden a una temperatura de 8 a 10 °C. Los resultados obtenidos muestran que la participación volumétrica máxima de racimo después de la destrucción por calentamiento preliminar constituye el 14% de GHWC a una temperatura de 13.4 °C. Cabe señalar una característica más; esta característica es adquirida por el agua debido a la presencia de gigantescos racimos. El agua que contiene GHWC puede considerarse un sistema disperso, en el que los grupos de agua juegan el papel de la fase dispersa. Las dimensiones de GHWC investigadas por nosotros estaban entre 2 y 40 μ y más. Tales dimensiones de las partículas son típicas de los sistemas dispersos. Una diferencia muy pequeña entre las densidades de la fase dispersa y el medio disperso conduce a la sedimentación y la estabilidad agregada de las suspensiones de racimo. Una de las características importantes del agua es la viscosidad. Se sabe que la dependencia de la temperatura de la viscosidad se describe mediante la ecuación exponencial y se muestra en la fig. 4.37a. Con base en los datos citados se encuentra la curva aproximada.
La naturaleza exponencial de la dependencia está determinada por el hecho de que en la interacción intermolecular, determinante de la viscosidad, participan los enlaces de hidrógeno. Dado que se observa una fuerte formación de grupos en el agua, fue interesante comparar las regularidades de temperatura de la formación de grupos y la viscosidad del agua. La figura 4.37b, c muestra la dependencia aproximada, obtenida por métodos matemáticos, para las curvas de calentamiento de agua pesada y destilada. De las curvas de diseño y las ecuaciones correspondientes a ellas se deduce que la regularidad de la variación del contenido de los grupos de GHWC para agua pesada y destilada está bien descrita por la dependencia exponencial. Esto significa que la destrucción de racimos al calentar agua está sujeta a regularidades generales del proceso tanto para el agua corriente como para el agua pesada. Tales regularidades incluyen la destrucción de la estructura de agua estructurada como hielo, la existencia de moléculas de agua libres (no unidas) de los huecos de la estructura similar al hielo y la formación como resultado de una masa uniforme no unida con GHWC. La diferencia entre agua destilada y agua pesada observada en el valor de los coeficientes en las ecuaciones de las curvas (Fig. 4.37b, c), indica una diferencia sustancial de las moléculas de agua que unen energía en el caso del enlace hidrógeno y deuterio. De la comparación de las ecuaciones de las curvas que se muestran en la fig. 3.37b, c con la ecuación de viscosidad (fig. 4.37a), se puede ver que los procesos bajo investigación están bien descritos por curvas exponenciales. Quizás los fenómenos que se observan constituyen regularidades similares, mientras que en la formación de cúmulos los enlaces de hidrógeno juegan un papel importante.
Así, este estudio sobre la física del agua utilizando métodos contemporáneos ha confirmado la posibilidad de la formación y existencia de conglomerados de diferentes dimensiones. Los efectos únicos de impacto físico utilizados en los procesos de tratamiento de agua abren nuevas vías para perfeccionar los procesos de preparación de agua potable de alta calidad. Los cambios en la composición estructural del agua, incluida la cantidad y concentración de grupos bajo los efectos externos, contienen información sobre la evolución de la atmósfera y la hidrosfera de la Tierra, que es necesaria para la estabilización del clima y la protección ecológica de la Tierra.
Referencias
[1] Fauque, Danielle. (2017). Joseph Priestley (1733-1804). Actualite Chimique. 60-61.
[2] Szostak, Rick. (2020). Industrial revolutions. 10.4324/9781003013518-30.
[3] Jack). Journal of Chemical Education - J CHEM EDUC. 49. 10.1021/ed049pA244.3.
[4] Zare, Richard. (2001). Book Review: Oxygen. By Carl Djerassi and Roald Hoffmann. Angewandte Chemie-international Edition - ANGEW CHEM INT ED. 40. 1971-1972. 10.1002/1521-3773(20010518)40:103.0.CO;2-N.
[5] Remane, Horst. (2010). Die Phlogistontheorie von Georg Ernst Stahl –. The Phlogiston Theory from Georg Ernst Stahl – Catalyst in the Development of Modern Scientific Chemistry. CHEMKON. 17. 10.1002/ckon.201010115.
[6] Nieddu, Martino. (2009). Bernadette Bensaude-Vincent et Jonathan Simon, 2008, Chemistry, The Impure Science, London, Imperial College Press, 268 p. + index.. Développement Durable et Territoir
[7] Quoted in the Encyclopaedia Britannica , 9th edition, vol. 5 (1876), 462.
[8] Kuhn, Thomas S. 1970. The structure of scientifi c revolutions , 2nd ed. Chicago: University of
Chicago Press.
[9] Klein, Ursula. (2021). Review of: Jungnickel, Christa and Russell McCormmach: Cavendish : the experimental life. Cranbury: Bucknell Univ. Press 1999. Ambix, v.48, 114-115 (2001).
[10] Klein, Ursula. (2004). Working and knowing in the history of STM. [Review of: Pickstone, John V. Ways of knowing. A new history of science, technology and medicine. Manchester, Manchester University Press, 2000].. Studies in history and philosophy of science. 35. 159-72.
[11] R, Cooper. (2002). Ways of Knowing: A New History of Science, Technology and Medicine. Social History of Medicine. 15. 162-163. 10.1093/shm/15.1.162.
[12] Ladyman, James. (2011). Structural Realism versus Standard Scientific Realism: The Case of Phlogiston and Dephlogisticated Air. Synthese. 180. 10.1007/s11229-009-9607-8.
[13] Servos, John & Ostwald, Wilhelm & Date, N.. (1983). Electrochemistry: History and Theory. Technology and Culture. 24. 125. 10.2307/3104182.
[14] Hentschel, Klaus. (1992). The uses of experiment, studies in the natural sciences, ed. by David Gooding ... : Cambridge ..., Cambridge University Press, 1989; [Rezension]. 10.18419/opus-7168.
[15] Lilly, M. (1988). The Sixth Sir Harold Hartley Memorial Lecture. Measurement and Control. 21. 100-107. 10.1177/002029408802100402.
[16] Banchetti, Marina. (2020). The Changing Relation between Atomicity and Elementarity: From Lavoisier to DaltonFrom Lavoisier to Dalton. 10.1093/oso/9780190933784.003.0006.
[17] Morrell, Jack. (1972). The Chemist Breeders: The Research Schools of Liebig and Thomas Thomson. Ambix. 19. 1-46. 10.1179/amb.1972.19.1.1.
[18] Mayo, Deborah. (2009). Toward progressive critical rationalism exchanges with Alan Musgrave. 113-124. 10.1017/CBO9780511657528.
[19] Sfetcu, Nicolae. (2020). Imre Lakatos, Preuves et Réfutations. 10.13140/RG.2.2.20798.31042.
[20] Mayo, Deborah. (2009). Toward progressive critical rationalism exchanges with Alan Musgrave. 113-124. 10.1017/CBO9780511657528.
[21] Odling, William. 1871. On the revived theory of phlogiston. Proceedings of the Royal Institution
of Great Britain 6: 315–325.
[22] Soler, Léna. 2008. Revealing the analytical structure and some intrinsic major diffi culties of the
contingentist/inevitabilist issue. Studies in History and Philosophy of Science 39: 230–241.
[23] Lashley, Conrad. (2020). The way things are, or are they?. 10.4324/9780367855383-12.
[24] Klein, Ursula. (2021). Review of: Chang, Hasok and Catherine Jackson (eds.) An element of controversy: the life of chlorine in science, technology and war. London: British Society for the History of Science 2007. The British Journal for the History of Science, v.42, 128?130 (2009).
[25] Schmidt, Jeremy. (2013). Hasok Chang: Is water H2O? Evidence, realism and pluralism. Water History. 5. 10.1007/s12685-013-0088-9.
[26] Nye, Mary. (2013). Is Water H2O? Evidence, Realism and Pluralism - by Hasok Chang. Centaurus. 55. 10.1111/1600-0498.12031.
[27] Gillies, Donald. (2008). HASOK CHANG Inventing Temperature: Measurement and Scientific Progress. British Journal for The Philosophy of Science. 60. 221-228. 10.1093/bjps/axn050.
[28] Brooks, Daniel. (2021). Adaptive Design, Contingency, and Ontological Principles for Limited Beings. Philosophy of Science.
[29] Larrauri Pertierra, Iñaki Xavier. (2020). Kuhn the Contextualist?. Aristos: A biannual journal featuring excellent student works. 5. 1-15. 10.32613/aristos/2020.5.1.4.
[30] Fuller, Steve. (2004). Historical Ontology by Ian Hacking. The Canadian Journal of Sociology / Cahiers canadiens de sociologie. 29. 478-479. 10.2307/3654682.
[31] Jones, David. (2002). Review: Ways of Knowing: A New History of Science, Technology and Medicine. Journal of the History of Medicine and Allied Sciences. 57. 10.1093/jhmas/57.2.239.
[32] Klein, Ursula. (2004). Working and knowing in the history of STM. [Review of: Pickstone, John V. Ways of knowing. A new history of science, technology and medicine. Manchester, Manchester University Press, 2000].. Studies in history and philosophy of science. 35. 159-72.
[33] Kauffman, George. (1994). Chemical Sciences in the Modern World (Mauskopf, Seymour H.). Journal of Chemical Education - J CHEM EDUC. 71. 10.1021/ed071pA311.
[34] Fleury-Heusghem, Nicole. (2000). Claude-Louis Berthollet (1748-1822). Bulletin de la Sabix. 5-12. 10.4000/sabix.239.
[35] Unruh, Jürgen. (2012). Luigi Valentino Brugnatelli - The first electroplater journeys into history - Part 4. Galvanotechnik. 103. 2172-2180.
[36] Siegel, Harvey. 1990. Educating reason: Rationality, critical thinking and education . New York:
Routledge.
[37] Le Grand, H.E. 1975. The “conversion” of C. L. Berthollet to Lavoisier’s chemistry. Ambix 22:
58–70.
[38] Liebig, Justus. 1851. Familiar letters on chemistry, in its relations to physiology, dietetics, agriculture, commerce, and political economy , 3rd ed. London: Taylor, Walton, & Maberly.
[39] Ghins, Michel. (2010). Bas van Fraassen on Scientific Representation. Analysis. 70. 524-536. 10.1093/analys/anq043.
[40] Simon, Jonathan. (2011). The Historiography of the Chemical Revolution: Patterns of Interpretation in the History of Science - by John McEvoy. Centaurus. 53. 10.1111/j.1600-0498.2010.00209.x.
[41] Klein, Ursula. (2005). ROBERT SIEGFRIED, From Elements to Atoms: A History of Chemical Composition. Transactions of the American Philosophical Society, 92 (4). Philadelphia: American Philosophical Society, 2002. Pp. x+278. ISBN 0-87169-924-9. No price given (paperback). British Journal for The History of Science. 38. 221-222. 10.1017/S0007087405226961.
[42] Simon, Jonathan. (2011). The Historiography of the Chemical Revolution: Patterns of Interpretation in the History of Science - by John McEvoy. Centaurus. 53. 10.1111/j.1600-0498.2010.00209.x.
[43] Allchin, Douglas. (1992). Phlogiston After Oxygen. Ambix. 39. 110-116. 10.1179/000269892790218610.
[44] Cunningham, Clifford. (2018). The Scientific Legacy of William Herschel. 10.1007/978-3-319-32826-3.
[45] Michael Faraday to Benjamin Abbott, 11 August 1812, in James ( 1991 ) , 17.
[46] Brooke, John. (1994). Book Review:The Fontana History of Chemistry William H. Brock. Isis. 85. 10.1086/356820.
[47] Kirwan, Richard. 1789. An essay on phlogiston and the composition of acids . London: J. Johnson.
[48] MORINI, SIMONA. (1996). M. NORTON WISE (a cura di), The Values of Precision, Princeton, Princeton University Press, 1995, vm + 372 pp.. Nuncius. 11. 385-388. 10.1163/182539196X01131.
[49] Gough, Jerry. (2004). Affinity, That Elusive Dream: A Genealogy of the Chemical Revolution (review). Technology and Culture - TECHNOL CULTURE. 45. 645-647. 10.1353/tech.2004.0129.
[50] Kim, Mi. (1996). Book Review:In the Shadow of Lavoisier: The Annales de Chimie and the Establishment of a New Science Maurice Crosland. Isis. 87. 10.1086/357618.
[51] Boantza, Victor. (2021). Styles of Experimental Reasoning in Early Modern Chemistry.
[52] Klein, Ursula. (2021). Review of: Fruton, Joseph S.: Methods and styles in the development of chemistry. Philadelphia: American Philosophical Society 2002. Ambix, v.50, 312-313 (2003).
[53] Joly, Bernard. (2014). Etienne-François Geoffroy (1672–1731), a Chemist on the Frontiers. Osiris. 29. 117-131. 10.1086/678100.
[54] Tang, Paul. (1991). Book Review:Die Wissenschaftsphilosophie Thomas S. Kuhns: Rekonstruktion und Grundlagenprobleme Paul Hoyningen-Huene. Isis. 82. 10.1086/356010.
[55] Hildebrand, Joel. (1947). Gilbert Newton Lewis. 1875-1946. Obituary Notices of Fellows of The Royal Society (1932-1954). 5. 491-506. 10.1098/rsbm.1947.0014.
[56] Allchin, Douglas. 1997. Rekindling phlogiston: From classroom case study to interdisciplinary relationships. Science and Education 6: 473–509.
[57] Mander, W.. (2020). Emotion and satisfaction in the philosophy of F. H. Bradley. 10.4324/9780429020223-3.
[58] Bachmann K (2008) Unidentified world: the Earth. GEO 1:76–94
[59] Mosin OV, et al (2015) Heavy Water as Medium for the Life of Organisms. Journal of Health, Medicine and Nursing. https://core.ac.uk/download/pdf/234691456.pdf
[60] Greene Brian (2020). Until the end of time. Mind, matter and our search for meaning in an evolving universe. Planeta: Barcelona.
[61] Albert Szent-Györgyi, «Biology and Pathology of Water», Perspectives in Biology and Medicine 14, n.º 2 (1971), p. 239.
[62] Andryushchenko, V & Boyko, E. (2020). Structuring of water molecules near a copper surface. Journal of Physics: Conference Series. 1677. 012122. 10.1088/1742-6596/1677/1/012122.
[63] Shu, Li & Jegatheesan, Leonardo & Jegatheesan, Veeriah & Li, Chun. (2020). The structure of water. Fluid Phase Equilibria. 511. 112514. 10.1016/j.fluid.2020.112514.
[64] Dore, John. (1990). Hydrogen-Bond Structures in Water & Ice. 10.1007/978-94-009-2157-3_15.
[65] Kucherenko, Natalia & Gladk?h, ?gor & Kapochkin, Borys & Kapochkina, Marharyta. (2017). Anomalous properties of water and their influence on hydroacoustic technologies. Bulletin of the National Technical University «KhPI» Series New solutions in modern technologies. 164-172. 10.20998/2413-4295.2017.07.23.
[66] Hoppmann, Mario & Richter, Maren & Smith, Inga & Jendersie, Stefan & Langhorne, Patricia & Thomas, David & Dieckmann, Gerhard. (2020). Platelet ice, the Southern Ocean's hidden ice: A review. Annals of Glaciology. 10.1017/aog.2020.54.
[67] Loerting, Thomas & Fuentes-Landete, Violeta & Handle, Philip & Seidl, Markus & Amann-Winkel, Katrin & Gainaru, Catalin & Böhmer, Roland. (2014). The glass transition in high-density amorphous ice. Journal of Non-Crystalline Solids. 407. 10.1016/j.jnoncrysol.2014.09.003.
[68] Pusztai, Laszlo. (2000). Structure of high- and low-density amorphous ice. Phys. Rev. B. 61. 10.1103/PhysRevB.61.28.
[69] Chen, Jing-Yin & Yoo, Choong-Shik. (2011). High density amorphous ice at room temperature. Proceedings of the National Academy of Sciences of the United States of America. 108. 7685-8. 10.1073/pnas.1100752108.
[70] Kucherenko, Natalia & Gladk?h, ?gor & Kapochkin, Borys & Kapochkina, Marharyta. (2017). Anomalous properties of water and their influence on hydroacoustic technologies. Bulletin of the National Technical University «KhPI» Series New solutions in modern technologies. 164-172. 10.20998/2413-4295.2017.07.23.
[71] Antonchenko VYa (1986) Fizika vody (Physics of water). Nauk Dumka, Kiev
[72] Kusalik, Peter & Svishchev, Igor. (1994). The Spatial Structure in Liquid Water. Science (New York, N.Y.). 265. 1219-21. 10.1126/science.265.5176.1219.
[73] Stoops, William. (2021). The Dielectric Polarization of Alcohols.
[74] Perry, Conaill & Zhang, Pengju & Nunes, Fernanda & Jordan, Inga & Conta, Aaron & Wörner, Hans Jakob. (2020). The Ionization Energy of Liquid Water Revisited. The Journal of Physical Chemistry Letters. XXXX. 10.1021/acs.jpclett.9b03391.
[75] Kiss, Péter & Baranyai, András. (2014). Anomalous properties of water predicted by the BK3 model. The Journal of Chemical Physics. 140. 10.1063/1.4871390.
[76] Folsome, Clair. (2021). Life : origin and evolution / with introductions by Clair Edwin Folsome. SERBIULA (sistema Librum 2.0).
[77] Grigorovich S (2004) In the beginning was RNA? Sci Life 2:44–55
[78] Dobretsov NL (2005) Early stages of life origin and evolution. Herald Vavilov Soc Genet
Breed Sci 9(1):43–54
[79] Perry, Conaill & Zhang, Pengju & Nunes, Fernanda & Jordan, Inga & Conta, Aaron & Wörner, Hans Jakob. (2020). The Ionization Energy of Liquid Water Revisited. The Journal of Physical Chemistry Letters. XXXX. 10.1021/acs.jpclett.9b03391.
[80] Scientific library (2007) http://sciencerussian.sibenzyme.com
[81] Rohilla, K. J., & Gagnon, K. T. (2017). RNA biology of disease-associated microsatellite repeat expansions. Acta neuropathologica communications, 5(1), 63. https://doi.org/10.1186/s40478-017-0468-y
[82] Meyer E. (1992). Internal water molecules and H-bonding in biological macromolecules: a review of structural features with functional implications. Protein science : a publication of the Protein Society, 1(12), 1543–1562. https://doi.org/10.1002/pro.5560011203
[83] Mulkidjanian A. Y. (2009). On the origin of life in the zinc world: 1. Photosynthesizing, porous edifices built of hydrothermally precipitated zinc sulfide as cradles of life on Earth. Biology direct, 4, 26. https://doi.org/10.1186/1745-6150-4-26
[84] Soper, AK. (2005). An asymmetric model for water structure. J. Phys.: Condens. Matter. 17. 10.1088/0953-8984/17/45/012.
[85] Kolyasnikov YuA (1993) Polytetrameric model of water structure. Herald Russ Acad Sci 63(8):730–732
[86] Morozov A. V. (2011). Sequence determinants of histone-DNA binding preferences: comment on "Cracking the chromatin code: precise rule of nucleosome positioning" by Edward N. Trifonov. Physics of life reviews, 8(1), 62–72. https://doi.org/10.1016/j.plrev.2011.01.013
[87] Averbakh, A. Z., Pekel, N. D., Seredenko, V. I., Kulikov, A. V., Gvozdev, R. I., & Rudakova, I. P. (1995). Flavin-dependent alcohol oxidase from the yeast Pichia pinus. Spatial localization of the coenzyme FAD in the protein structure: hot-tritium bombardment and ESR experiments. The Biochemical journal, 310 ( Pt 2)(Pt 2), 601–604. https://doi.org/10.1042/bj3100601
[88] Nielsen, Jonathan & Dyrby, Tim & Mathiesen, Joachim & Lundell, Henrik. (2015). Probing the fractal structure of grey matter dendrite networks using high frequency oscillating gradient diffusion weighted MRI.
[89] Angell CA, Franks F (ed) (1981) Water: a comprehensive treatise, in water and aqueous solutions
at subzero temperatures. Plenum Press, New York
[90] Romankevich, E. & Vetrov, A.. (2013). Masses of carbon in the Earth’s hydrosphere. Geochemistry International. 51. 10.1134/S0016702913060062.
[91] Brickwedde, Ferdinand. (1982). Harold Urey and the discovery of deuterium. Physics Today - PHYS TODAY. 35. 34-42. 10.1063/1.2915259.
[92] Stanojevic, Nenad & Djokic, Jelena & Osmokrovic, Predrag. (2019). Research on water interconnections within the Sar Mountains aquatorium by radioactive hydrogen isotope tritium. Nuclear Technology and Radiation Protection. 34. 40-40. 10.2298/NTRP191029040S.
[93] Timakov AA (2003) Main effects of light water. In: Abstract of the 8th Russian international scientific conference of Physical-chemical processes during selection of atoms and molecules, RSC Kurchatov Institute, Moscow, 6–10 Nov 2003
[94] Nakahara, Masaru & Matubayasi, Nobuyuki. (1999). Physical properties and structure of supercritical water. Electrochemistry -Tokyo-. 67. 988-993. 10.5796/electrochemistry.67.988.
[95] Varnavsky IN (2000) New technology and equipment for production purified biologically active healing drinking water. Dissertation, Moscow Institute of Medical and Biological Problems
[96] Watson JD, Crick FHC (1953) Genetical implication of the structure of deoxyribose nucleic acid. Nature 17:964–966
[97] Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999 Dec 3;286(5446):1897-905. doi: 10.1126/science.286.5446.1897. PMID: 10583946.
[98] Lamprecht J, Schroeter D, Paweletz N. Derangement of microtubule arrays in interphase and mitotic PtK2 cells treated with deuterium oxide (heavy water). J Cell Sci. 1991 Apr;98 ( Pt 4):463-73. PMID: 1650376.
[99] Yakhno, Tatiana & Drozdov, Mikhail & Yakhno, Vladimir. (2019). Giant Water Clusters: Where Are They From?. International Journal of Molecular Sciences. 20. 1-12.
[100] Basov, A., Fedulova, L., Baryshev, M., & Dzhimak, S. (2019). Deuterium-Depleted Water Influence on the Isotope 2H/1H Regulation in Body and Individual Adaptation. Nutrients, 11(8), 1903. https://doi.org/10.3390/nu11081903
[101] Goodall, Kirk. (2021). In Search of the Fountain of Youth Preliminary Analysis of Deuterium's Role in DNA Degradation.
[102] Goodall KB (2003) In search of the fountain of youth. Preliminary analysis of deuterium’s role in DNA degradation. Anti-aging medical news: the official newsletter of the American Academy of Anti-Aging Medicine 7–31
[103] Boukar, Ousman & Fifen, Jean & Malloum, Alhadji & Zoubeida, Dhaouadi & Ghalila, Hassen & Conradie, Jeanet. (2019). Structures of Solvated Ferrous Ion Clusters in ammonia and Spin-crossover at various Temperatures. New Journal of Chemistry. 43. 10.1039/C9NJ02462J.
[104] Mi, Dongbo & Chingin, Konstantin. (2020). Water Radical Cations in the Gas Phase: Methods and Mechanisms of Formation, Structure and Chemical Properties. Molecules. 25. 3490. 10.3390/molecules25153490.
[105] Antonchenko VY, Davydov AS, Ilyin VV (1991) Osnovy fiziki vody (Fundamentals of physics
of water). Nauk Dumka, Kiev
[106] Antonchenko VYa, Davydov AS, Zolotariuk AV (1985) Solution and proton motion in icelike
structures. Phys Stat Sol (B) 115(2):631–640
[107] Wilson KR, Cavalleri M, Rude BS et al (2000) Characterization of hydrogen bond acceptor molecules at the water surface using near-edge X-ray absorption fine-structure spectroscopy
and density functional theory. J Phys Condens Matter 14(8):L221–L226
[108] Sun, Zhaoru & Zheng, Lixin & Chen, Mohan & Klein, Michael & Paesani, Francesco & Xifan, Wu. (2018). Electron-Hole Theory of the Effect of Quantum Nuclei on the X-Ray Absorption Spectra of Liquid Water. Physical Review Letters. 121. 10.1103/PhysRevLett.121.137401.
[109] Vuks MF (1989) Dielectric polarization alcohol in solutions. Res Water Aqueous Syst Phys
Meth 73:172–180
[110] Zhukovsky AP (1981) Motivation of the continuum model of water structure by IR spectroscopy. J Struct Chem 22:56–63
[111] Yu, Xiaoxuan & Tang, Yao & Pan, Jian & Shen, Lin & Begum, Afruza & Gong, Zhanyang & Xue, Jinkai. (2020). Physico-Chemical Processes. Water Environment Research. 92. 10.1002/wer.1430.
[112] Zatsepina GN (1998) Fizicheskiye svoystva i struktura vody (Physical properties and structure
of water). Izd-vo MGU, Moscow
[113] https://courses.lumenlearning.com/introchem/chapter/the-structure-and-properties-of-water/
[114] https://ergodic.ugr.es/termo/lecciones/water2.html
[115] Daschbach JL, Peden BM, Smith RS et al (2004) Adsorption, desorption, and clustering H2O on Pt (III). J Chem Phys 120(3):1516–1523
[116] Fajardo ME, Tam S (2001) Observation of the cyclic water hexamer in solid parahydrogen. J Chem Phys 115(15):6807–6810
[117] Anglada, Josep & Hoffman, Gerald & Slipchenko, Lyudmila & Martins-Costa, Marilia & Ruiz-Lopez, Manuel & Francisco, Joseph. (2013). The Atmospheric Significance of Water Clusters and Ozone-Water Complexes.. The journal of physical chemistry. A. 117. 10.1021/jp407282c.
[118] Wierzejewska, Maria & Sompolski, Daniel. (2008). Ab initio MP2 and FTIR matrix isolation studies on tert-butanethiol complexes with water. Journal of Molecular Structure - J MOL STRUCT. 872. 166-175. 10.1016/j.molstruc.2007.02.037.
[119] Kamberaj, Hiqmet. (2020). Molecular Dynamics Methods in Simulations of Macromolecules. 10.1007/978-3-030-35702-3_6.
[120] Kamberaj, Hiqmet. (2016). Advanced Molecular Dynamics Methods.
[121] Hahalin ?V (2006) The influence of a low-intensive electromagnetic field on water clusters in the presence of ions. Abst of Ph.Sc Thesis, Moscow State University.
[122] CNews R&D, Natural Science (2010) http://rnd.cnews.ru/naturs_cience/news/top/index_science.
shtml.
[123] Goncharuk VV, Kamalov GL, Kovtun G? et al (2002) Kataliz. Mekhanizmy gomogennogo igeterogennogo kataliza, klasternyye podkhody (Catalysis: mechanisms of homogeneous and
heterogeneous catalysis, cluster approaches). Nauk Dumka, Kiev
[124] Goncharuk, V. & Smirnov, V. & Syroyeshkin, A. & Malyarenko, V.. (2007). Clusters and gigantic heterophase water clusters. Journal of Water Chemistry and Technology. 29. 1-8. 10.3103/S1063455X07010018.
[125] Bernatskiy, Anton & Lagunov, Vladimir & Ochkin, V.. (2019). Investigation of the Concentration Dynamics of H2O and HDO Molecules in a Discharge. Physics of Atomic Nuclei. 82. 1382-1386. 10.1134/S1063778819100065.
[126] Frenkel J (1946) Kinetic theory of liquids. Oxford University Press, Oxford
[127] Morrison SR (1977) The chemical physics of surfaces. Plenum Press, London
[128] Mishchenko KP, Poltoratsky G? (1974) Termodinamika i stroyeniye vodnykh i nevodnykh astvorov elektrolitov (Thermodynamics and structure of aqueous and nanoqueous solution
of electrolytes). Khimiya, Leningrad
[129] Holloway MA, Holloway JW (2003) Micro-cluster liquids and methods of making and using
them.
[130] Bogatin JG, Podoyma V (1999) Magnetohydrodynamic device. US Patent 5866010,
[131] Timakov ?? (2003) Main effects of light water. In abstract of the 8-th Russian international scientific conference of “Physical-chemical processes during selection of atoms and molecules”,
RSC Kurchatov Institute, Moscow, 6–10 Nov 2003
[132] Dinu, Dennis & Podewitz, Maren & Grothe, Hinrich & Loerting, Thomas & Liedl, Klaus. (2020). On the synergy of matrix-isolation infrared spectroscopy and vibrational configuration interaction computations. Theoretica Chimica Acta. 10.1007/s00214-020-02682-0.
[133] Ivanov, Evgeniy & Yu, Elena & Lebedeva, & Abrosimov, Vladimir & Nadezhda, · & Ivanova, G. (2013). Densimetric Studies of Binary Solutions Involving H 2 O or D 2 O as a Solute in Dimethylsulfoxide at Temperatures from (293.15 to 328.15) K and Atmospheric Pressure. Journal of Solution Chemistry. 41. 3311-3333. 10.1007/s10953-012-9877-5.
[134] Ignatov, Ignat & Mosin, O.. (2013). Structural Mathematical Models Describing Water Clusters. Journal of Mathematical Theory and Modeling. 3. 72-87. 10.13187/ejnr.2014.3.141.
[135] Schaftenaar, Gijs & Noordik, J.. (2000). Molden: A Pre- and Post-Processing Program for Molecular and Electronic Structures. Journal of computer-aided molecular design. 14. 123-34. 10.1023/A:1008193805436.
[136] Hasted JB (1972) Liquid water: dielectric properties. Plenum Press, New York
[137] Fomey D, Jacox ME, Thompson WE (1993) The mid- and near-infrared spectra of water and water dimer isolated in solid neon. J Mol Spectrosc 157(2):479–493
[138] Dyke TR, Mack KM, Muenter JS (1977) The structure of water dimer from molecular beam electric resonance spectroscopy. J Chem Phys 66(2):498–510
[139] Huang ZS, Miller RE (1989) High-resolution near-infrared spectroscopy of water dimer. J Chem Phys 91(11):6613–6631
[140] Ch'ng, Lee & Samanta, Amit & Wang, Yimin & Bowman, Joel & Reisler, Hanna. (2013). Experimental and Theoretical Investigations of the Dissociation Energy (D-0) and Dynamics of the Water Trimer, (H2O)(3). The journal of physical chemistry. A. 117. 10.1021/jp401155v.
[141] Perez-Torres, Jhon & Hadad, Cacier & Restrepo, Albeiro. (2008). Structural studies of the water tetramer. International Journal of Quantum Chemistry. 108. 1653 - 1659. 10.1002/qua.21615.
[142] Balasubramanian, K.. (2004). Nonrigid Group Theory, Tunneling Splittings, and Nuclear Spin Statistics of Water Pentamer: (H 2 O) 5. Journal of Physical Chemistry A - J PHYS CHEM A. 108. 5527-5536. 10.1021/jp049955k.
[143] Chen, Yiming & Li, Hui. (2010). Intermolecular Interaction in Water Hexamer. The journal of physical chemistry. A. 114. 11719-24. 10.1021/jp104822e.
[144] Feynman R, Leighton R, Sands ? (1977) Electricity and magnetism. Addison Wesley Publish
Company, Boston
[145] Higashitani K, Oshitani J (1997) Measurements of magnetic effects on electrolyte solutions
by atomic force microscope. Proc Safety Environ Prot 75(2):115–119
[146] Brovchenko I, Paschek D, Geiger A (2000) Gibbs ensemble simulation of water in spherical
cavities. J Chem Phys 113(22):5026–5036
[147] Bernal JD (1956) The geometric constructions with water molecules. Russ Chem Rev 25:643–660
[148] Drost-Hansen W, Clegg J (1979) Cell-associated water. Academic Press, London
[149] Sastry S (2001) Water structure: order and oddities (news and views). Nature 409(1):300–301
[150] Sastry S (2001) Water structure: order and oddities (news and views). Nature 409(1):300–301
[151] Liu K, Cruzan JD, Saykally RJ (1996) Water clusters. Science 3:929–993
[152] Morkovnik ?F, Okhlobystin ?Yu (1979) Inorganic radical-ions and their organic reactions. Russ Chem Rev 48(11):1968–2006
[153] Swallow AJ (1973) Radiation chemistry. Longman, London
[154] Malyarenko VV (1993) Nature of free radicals of humine acids. J Water Chem Technol 15(9/10):611–620
[155] Rozantsev EG, Sholle VD (1979) Organicheskaya khimiya svobodnykh radikalov (Organic chemistry of free radicals). Khimiya, ?oscow
[156] Knorre DG, Krylova LF, Muzykantov VS (1990) Fizicheskaya khimiya (Physical chemistry). Vyssh Shkola, Moscow
[157] Borisova ?E, Koikov SN (1979) Fizika dielektrolitov (Physics of dielectric). Izd-vo Leningr un-ta, Leningrad
[158] Goncharuk VV, Malyarenko VV (2007) The study of sound absorbtion in water. J Water Chem Technol 29(2):65–71
[159] Mason WP (1966) Physical acoustics. Academic Press, New York
[160] Rozenberg LD (1968) Moshchnyye ul’trazvukovyye polya (Powerful ultrasonic fields). Nauka, Moscow
[161] Mishchenko KP, Poltoratsky G? (1974) Termodinamika i stroyeniye vodnykh i nevodnykh rastvorov elektrolitov (Thermodynamics and structure of aqueous and nanoqueous solution of electrolytes). Khimiya, Leningrad
[162] Goncharuk VV, Malyarenko VV (2001) Changes in properties of water affected by electrochemical treatment. J Water Chem Technol 23(4):345–353
[163] Qu KY, Jiang Y (2000) Studies on randomness and nucleation rate of freezing from homogeneous
nucleation. Acta Phys Sin 49(11):2214–2219
[164] Kumar, Pradeep & Sathyamurthy, Narayanasami. (2018). An ab initio quantum chemical investigation of the structure and stability of ozone-water complexes. Chemical Physics. 415.
[165] Nelson DD, Zahniser MS (1994) A mechanistic study of the reaction of HO2 radical with ozone. J Phys Chem 98(8):2101–2104
[166] Gillies JZ, Gillies CW, Suenram RD et al (1991) A microwave spectral and Ab Initio investigation of O3-H2O. J Mol Spectrosc 146:493–512
[167] Zakharov II, Kolbasina OI, Semenyuk TN ?t al (1993) Molecular ozone-water complex. Ab initio calculation with inclusion of electron correlation. J Str Chem 34(3):359–362
[168] Tachikawa H, Abe S (2003) Ozone-water 1:1 complexes O3-H2O: An ab initio study. Inorg Chem 42:2188–2190
[169] Schaftenaar G, Noordik JH (2000) Molden: a pre- and post-processing program for molecular
and electronic structures. J Comput-Aided Mol Design 14:123–134
[170] Loboda AA, Goncharuk VV (2009) Ab Initio calculation of water clusters. The vibratory analysis and isotopic effect. J Water Chem Technol 31(2):98–109
[171] Tachikawa H, Abe S (2003) Ozone-water 1:1 complexes O3-H2O: An ab initio study. Inorg Chem 42:2188–2190
[172] Mishchenko KP, Poltoratsky G? (1974) Termodinamika i stroyeniye vodnykh i nevodnykh rastvorov elektrolitov (Thermodynamics and structure of aqueous and nanoqueous solution
of electrolytes). Khimiya, Leningrad
[173] Knorre DG, Krylova LF, Muzykantov VS (1990) Fizicheskaya khimiya (Physical chemistry). Vyssh Shkola, Moscow
[174] Malyarenko VV (1989) Thermodynamic assessment of the temperature of supercooled water,
based on two-structure mode. Ukr Chem J 55(8):810–814
[175] Chaplin MF (2006) Water structuring at colloidal surfaces. In: Blitz J, Gun’ko V (eds) Surface
chemistry in biomedical and environmental science. NATO security through science
series, Springer, The Netherlands
[176] Goncharuk VV, ?rehova L?, Malyarenko VV (2008) Influence of temperature on water clusters. J Water Chem Technol 30(2):80–84
[177] Goncharuk VV, Bulanova AV, Malyarenko VV (2006) The water as a heterogeneous cluster
disperse system. Paper presented at the X Ukrainian-Polish symposium theoretical and
experimental studies of interfacial phenomena and their technological applications. Lviv—
Uslissia, 26–30 Sept 2006
[178] Knorre DG, Krylova LF, Muzykantov VS (1990) Fizicheskaya khimiya (Physical chemistry). Vyssh Shkola, Moscow
Autores:
Eduardo Ochoa Hernández
Nicolás Zamudio Hernández
Abraham Zamudio Durán
Lizbeth Guadalupe Villalon Magallan
Mónica Rico Reyes
Pedro Gallegos Facio
Gerardo Sánchez Fernández
Rogelio Ochoa Barragán