Texto académico
Autores
Eduardo Ochoa Hernández
Nicolás Zamudio Hernández
Filo Enrique Borjas García
Lizbeth Guadalupe Villalon Magallan
Pedro Gallegos Facio
Gerardo Sánchez Fernández
Rogelio Ochoa Barragán

Módulo 3. Teoría Celular
3.1 ¿Cuándo?
Aproximadamente 250 años antes de la definición de célula de Sachs, Robert Hooke observó células por primera vez en corcho. Las observaciones de Hooke no solo iniciaron una nueva ola en el estudio de la biología vegetal, sino que también nos dieron el término "célula[1]". Las raíces etimológicas del término se encuentran en la palabra latina cellulae, que significa celdas hexagonales del panal[2]. Poco después de que Hooke hiciera sus observaciones y acuñara el término "célula", Antony van Leeuwenhoek descubrió microorganismos móviles[3]. Más tarde, Marcello Malpighi y Nehemiah Grew publicaron observaciones detalladas de los diferentes órganos y tejidos de las plantas[4]. Grew describió las celdas en forma de panal, pero también otras formas de celdas, que formaban la corteza y la médula (Grew 1682). La biología celular ha avanzado mucho desde la época de Hooke. Con los avances en microscopía[5], se ha vuelto más fácil observar otros organelos y estructuras como paredes celulares, núcleos y cloroplastos, y comprender los mecanismos de división celular y otros procesos. De hecho, se puede decir que la pared celular es el primer componente de la célula que se observó cuando Hooke miró las células de corcho. Se puede decir que el siglo XIX es el siglo de la biología celular. El descubrimiento del núcleo, las mitocondrias, el protoplasma, el Golgi, etc. y los fundamentos de la teoría celular ocurrieron en el siglo XIX.
Estudiando las hojas de las orquídeas, Robert Brown descubrió el núcleo, que definió así: “En cada célula de la epidermis de gran parte de esta familia, especialmente de las que tienen hojas membranáceas, una singular areola circular, generalmente algo más opaca que la la membrana de la célula es observable. Esta areola [núcleo], que es más o menos claramente granular, es ligeramente convexa y, aunque parece estar en la superficie, en realidad está cubierta por la lámina exterior de la célula[6]”. El conocimiento de la estructura y la biología del núcleo allanó el camino para una mejor comprensión de la genética y, por lo tanto, ayudó en ciencias aplicadas como el mejoramiento de cultivos.
El descubrimiento del núcleo fue seguido por la formulación de la teoría celular por Matthias Jacob Schleiden y Theodor Schwann[7]. Hugo von Mohl (1852) dio el término "protoplasma" y describió el movimiento de la savia celular[8]. El protoplasma según lo definido por von Mohl es "líquido opaco, viscoso de color blanco, con gránulos entremezclados". Los gránulos a los que se refiere son orgánulos como plástidos, mitocondrias y Golgi[9]. Pasaron más de 300 años después del descubrimiento de las células (Hooke 1665) para aceptar la naturaleza endosimbiótica de las células eucariotas[10]. En biología celular, los orgelos o estructuras a menudo se informaron mucho antes de que se estudiaran en detalle. Bohumil Nemec descubrió filamentos gruesos que recorrían longitudinalmente los bordes de las células en los ápices de las raíces[11], que luego se demostró que eran haces de actina F que se extendían a través de plasmodesmas[12].
El descubrimiento de los cloroplastos fue similar. En una nota general, los plástidos eran conocidos por los antiguos indios[13]. Wilhelm Hofmeister observó la placa celular en las células vegetales en división ya en 1867, cuando la división de células eucariotas no se entendía bien[14].
3.2 ¿Cómo?
Anthony Leeuwenhoek descubrió por primera vez las maravillas del mundo viviente, que antes que él eran invisibles[15]. Los avances en el desarrollo de lentes superiores de Abbe y Schott en Alemania llevaron las observaciones a un mayor nivel de aumento[16]. La importancia de la microscopía para la fisiología fue sugerida por Jan Evangelista Purkinje en la primera mitad del siglo XIX[17]. Hemos recorrido un largo camino desde las primeras observaciones de la célula por Hooke hasta el estudio detallado de los organelos posibles hoy en día utilizando los conceptos de microscopía confocal. Con la microscopía avanzada, los avances en colorantes y sondas también han jugado un papel importante al permitir el estudio detallado de las células y sus componentes. Los avances en microscopía nos han permitido no solo ver los organelos, sino también observar y comprender las interacciones entre ellos. Además de los avances en microscopía, microtomía e histoquímica, la capacidad de hacer crecer las células ha permitido un rápido progreso en nuestra comprensión de las células vegetales. A Haberlandt (1902) se le atribuye ser el primero en cultivar células vegetales en soluciones nutritivas[18]. Vasil (2008) revisó la historia del cultivo de células vegetales y su uso en biotecnología[19]. Los parámetros celulares como el tamaño, la forma y el número pueden estudiarse en cultivos celulares y usarse para el fenotipado celular[20].
3.3 ¿Por qué?
La biología celular a veces se considera una ciencia básica, lo que no es cierto debido a las implicaciones que tiene en campos aplicados como la medicina y la agricultura[21]. El estudio de las células vegetales no solo ha mejorado nuestra comprensión de las plantas, sino que también ha facilitado el aprovechamiento de los recursos vegetales para fines como la biotecnología de cultivos. Cómo podemos llevar la biología celular a un nuevo nivel integrando el conocimiento conceptual de los biólogos celulares (botánicos) del siglo XIX y las herramientas modernas de alto rendimiento. Otro aspecto importante es que los conceptos científicos son muy importantes para la interpretación adecuada de los datos obtenidos y aún más importante para hacer las preguntas correctas y elegir una metodología de investigación y un diseño experimental adecuados. Al guiar la llamada ciencia normal, los conceptos científicos dan forma a paradigmas emergentes y maduros hasta que los nuevos conceptos conducen al derrocamiento revolucionario del viejo paradigma y plantean nuevos paradigmas emergentes[22]. Por ejemplo, el concepto de generación espontánea de vida fue propuesto por Aristóteles y dominó nuestro pensamiento sobre la vida hasta que Louis Pasteur refutó esta teoría[23]. Recientemente, ha habido varios cambios de paradigma en genética, ciencias de las plantas y neurociencias[24]; teoría de la evolución[25]; y en el pilar básico de la biología celular, la teoría celular[26]. Primeros intentos de comprender la naturaleza de la conciencia y sus roles en biología[27] indican que un nuevo cambio de paradigma es inminente. Muchos descubrimientos cruciales en biología celular se lograron mediante el estudio de las plantas, incluidos los descubrimientos de la célula, el núcleo, el origen simbiótico de orgánulos eucariotas, microtúbulos, canales célula-célula, cromosomas, mitosis y ciclo celular[28]. Dado que las plantas superiores están demostrando ser organismos conductualmente activos y cognitivos[29], y su comportamiento activo sea sensible a los anestésicos[30], es posible que las plantas también resulten cruciales para nuestra comprensión de la naturaleza elusiva de la conciencia.
3.4 Fundamentos teóricos y metódicos de la célula
Varias teorías, primero en forma de narrativas mitológicas, sobre los orígenes y la base de la vida han existido desde los albores de la humanidad. Algunas de las primeras ideas modernas sobre la sustancia de la vida y su programa de desarrollo confinado a una pequeña parte de un organismo vivo provienen de Aristóteles en el siglo IV a. C. Sobre la base de la experiencia empírica del desarrollo del huevo y la reproducción vegetativa de las plantas, postuló la "entelquía" como un principio impulsor que lleva a los organismos hacia la realización de su forma y potencial[31]. Hasta el siglo XVII, el desarrollo de la ciencia de la biología raras veces buscó las causas de los fenómenos vivos en la fina estructura de los organismos. Tendencias prominentes como la morfología francesa y la Naturphilosophie alemana buscaron una explicación de los planes y estructuras corporales en formas ideales abstractas hacia las que se dirigen los organismos[32]; el enfoque fue en gran parte ortogonal a las visiones mecanicistas posteriores y contemporáneas de la vida. Los conceptos idealistas acompañaron aún más a la biología, pero en el siglo XVIII apareció un cambio gradual hacia el empirismo y las tendencias mecanicistas de la ciencia. La tendencia incluyó el resurgimiento del atomismo, una teoría que afirma que las propiedades de la materia vienen dadas por las pequeñas partículas indivisibles de las que está compuesta[33]. Es importante señalar que los avances que dieron como resultado la formulación de la teoría celular no solo fueron liderados por mejoras tecnológicas en microscopía, sino también por un cambio en el enfoque teórico. Muchos estudiosos ya tenían la idea de que los tejidos observados eran agregados de unidades más básicas, incluso antes de mirar a través de microscopios. Otra inspiración filosófica importante (bastante distinta de las primeras analogías comunes entre células y átomos o cristales, y mucho más cercana a la percepción contemporánea de las células) vino de G.W. Leibniz (1646-1716). Su idea estableció la base, a menudo desconocida, de la teoría celular. En la idea de "mónadas" autorreproductoras totalmente autónomas, desarrollada en un discurso crítico con la visión mecanicista cartesiana del universo, Leibniz afirmó que si los organismos vivos fueran máquinas, sus partes no serían simplemente piezas mecánicas de materia, sino máquinas más pequeñas. Es importante destacar que la dinámica de las mónadas se impulsa desde el interior. Esta idea estimuló el concepto del filósofo alemán Lorenz Oken (1779-1851) de que todos los organismos están compuestos de “infusorios” y “Urbla¨schen” (burbujas primordiales) como unidades básicas de vida; esta especulación precedió directamente a los trabajos de los primeros biólogos celulares empíricos[34]. Sin embargo, fue solo la invención y la mejora de los microscopios lo que permitió la observación directa de la base material y la composición de los organismos. Sobre la base de las primeras observaciones, se postuló la composición de los tejidos como fibras, glóbulos o cilindros retorcidos.
En el siglo XVIII, Albrecht von Haller, inspirado por el atomismo, especuló que las fibras compuestas por cadenas de átomos eran los elementos estructurales básicos del cuerpo: “Para el fisiólogo la fibra es lo que la línea recta para el geómetra, y de esta fibra todas las formas surgen seguramente”. Robert Hooke estuvo activo en muchos campos de las ciencias naturales en la segunda mitad del siglo XVII y se le considera el padre del término "célula" en biología. Usó este término para describir las estructuras que vio con su microscopio simple en rodajas de tejido de corcho vegetal porque se parecían a células de panal (celulas en latín). En ese momento, las células fueron concebidas como huecas y consideradas como "avenidas de comunicación, canales para la transmisión de jugos[35]".
3.5 De Schleiden a Virchow: formación de la teoría celular
Principios cada vez más científicos del siglo XIX estaban convencidos de que los tejidos vegetales generalmente estaban compuestos de células, pero Matthias Jakob Schleiden (1804-1881) hizo el primer intento de utilizar la composición celular como un principio explicativo unificador en botánica. Schleiden quería establecer la botánica sobre una base firme como una ciencia más exacta, dejando atrás la tradición especulativa de la Naturphilosophie alemana. Schleiden tenía una orientación mecanicista y, como muchos de sus contemporáneos, se inspiró en la física de Isaac Newton[36]. Usó metáforas cristalinas para conceptualizar la autoorganización de los organismos. También enfatizó los enfoques inductivos y empíricos, así como la importancia de seguir la ontogenia (que refleja la especificación o diferenciación de formas generales iniciales más simples en formas elaboradas más complejas) para comprender adecuadamente los tejidos vegetales. Los esfuerzos de Schleiden dieron como resultado la formulación de una regla general de que todos los tejidos vegetales están compuestos por un solo elemento básico, la célula poliédrica. Posteriormente, pediría "la condena de toda teoría que explica los procesos en una planta de otra manera que como una combinación de procesos en células individuales". La pared celular como elemento de contorno y estructura todavía se consideraba más importante que el contenido interno, aunque el núcleo ya era conocido y descrito. Fue nombrado en 1833 por Robert Brown quien, sin embargo, no reconoció la presencia general de núcleos en todas las células. Tal opinión es comprensible considerando que la pared celular es morfológicamente la estructura más conspicua en las células vegetales diferenciadas y, a menudo, es el determinante funcional del tejido particular. Además, la importancia crucial de la mecánica de la pared celular y su integración con la membrana de la célula vegetal y el núcleo citoplásmico son características bien reconocidas en la actualidad de la organización del cuerpo vegetal. Schleiden no tenía claro el origen ontogénico de las células; por lo tanto, un protoplasma extracelular o savia jugó un papel en su concepto de formación celular. Postuló la condensación de núcleos a partir de este material y la formación de materia celular a su alrededor. Las células se formaron a partir de núcleos como vesículas en crecimiento hasta que se tocaron entre sí. Más adelante en el desarrollo, se formaron principalmente alrededor de núcleos dentro de otras células[37]. Schleiden consideró que los núcleos de las células maduras eran prescindibles y, a menudo, se reabsorbían. Debido a la ausencia de paredes celulares distintas y las dificultades en la preparación de la muestra, la naturaleza celular de los cuerpos de los animales fue menos clara. Se estudiaron células animales, por ejemplo, en embriones en desarrollo. Sin embargo, la suposición empírica general postulaba la formación del cuerpo animal a partir de células durante el desarrollo temprano, pero no necesariamente en su estado adulto. Henri Dutrochet (1776-1847) defendía una cosmovisión materialista y tenía como objetivo identificar fenómenos vitales en animales y plantas.
Afirmó que tanto los tejidos vegetales como los animales estaban compuestos de "vesículas" y "glóbulos", aunque probablemente no pudo observar las células animales. Aunque la visión morfológica de las células de Dutrochet era en gran medida errónea, probablemente fue el primero en percibir las células como unidades fisiológicas básicas de metabolismo intercambio con entrada selectiva de nutrientes y salida de residuos. También sugirió la existencia de los mismos principios subyacentes en los tejidos animales y vegetales: “[Si] se rastrean los fenómenos hasta sus orígenes, se ve que las diferencias desaparecen y se revela una admirable uniformidad de plan”. La primera afirmación de la presencia generalizada de "Kornchen" análogo a las células vegetales en los tejidos animales, respaldada por innumerables observaciones histológicas, fue realizada por Bohemiam Jan Evangelista[38]. Afirmó que los tejidos animales estaban compuestos universalmente por células, fibras y fluidos. Purkyne también fue uno de los primeros (después de Dutrochet) en enfatizar el significado funcional de las células y facilitó la transición de la “histomorfología” a la “histo-fisiología”, particularmente a través de estudios completos de los movimientos ciliares en varios tejidos animales. A diferencia de Schwann, que puso mayor énfasis en el núcleo, Purkyne también se centró en el contenido activo de la célula, el "protoplasma".
Theodor Schwann (1810-1882) obtuvo el mayor crédito por extender la teoría celular al tejido animal porque hizo afirmaciones más fuertes (aunque no siempre correctas) que Purkyne. Inspirado por las conclusiones de Schleiden, así como por la similitud entre las células de notocorda animal y las células vegetales descubiertas por el maestro de Schwann, Johannes Müller, Schwann acumuló una gran cantidad de ejemplos de tejidos animales embrionarios y adultos que consisten en células y afirmó que el origen celular es el principio ontogénico unificador tanto para animales como para plantas[39]. Schwann no estaba seguro sobre el origen exacto de las células individuales y postuló su origen ya sea a partir de materia de vida homogénea (posiblemente a través de la generación inicial de un núcleo) o desde el interior de otras células, alrededor de sus núcleos. Según Schwann, las células podrían originarse dentro o fuera de otras células.
Inspirado por Schleiden, Schwann afirmó que la formación de células a partir de líquido a través de núcleos era un proceso mecanicista similar a la cristalización. Coexistieron varias ideas diferentes sobre el mecanismo de generación de nuevas células y muchos científicos aceptaron que diferentes mecanismos podrían funcionar en diferentes organismos y tejidos. El descubrimiento de la fisión de células binarias por Barthelemy Dumortier y Hugo von Mohl fue de gran importancia, aunque ambos admitieron la pluralidad de mecanismos de formación de células. Franz Unger (1800-1870) fue el primero en oponerse abiertamente a la idea de agregación / cristalización de Schleiden. Descartó los "citoblastos" como fuente de células y postuló que la división binaria era el mecanismo más común de división de células vegetales (Harris 2000). En unos pocos años, se había acumulado suficiente evidencia empírica para abandonar el concepto de formación celular de Schleiden. Debido a dificultades técnicas, tomó mucho más tiempo acumular observaciones precisas de la formación de células animales. Robert Remak (1815-1865) propuso la primera teoría unificadora explícita de la división celular tanto en plantas como en animales. Remak desarrolló nuevos agentes endurecedores que le permitieron llevar a cabo estudios exhaustivos sobre la formación de células en muchos tejidos animales. Concluyó que la formación extracelular de células no ocurre en tejidos animales y que la división binaria es el mecanismo universal de formación celular. El desarrollo es, pues, una secuencia de divisiones binarias seguida de modificaciones morfológicas; además, el huevo en sí es una célula. Remak también propuso que las mismas reglas regían la celda división en tejidos tanto patológicos como embrionarios (en oposición directa a la teoría de Müller de la formación de tumores malignos específicos). Remak se opuso categóricamente a Schleiden y Schwann, en particular a sus analogías entre células y cristales: “No es necesario hacer una mención especial a la similitud o disparidad de células y cristales, porque, a la luz de los hechos que he discutido, las dos estructuras no ofrecen puntos de comparación”. Rudolf Virchow (1821-1902), fuertemente inspirado por Remak y difundiendo las ideas de Remak, consolidó la teoría celular con su famosa declaración "Omnis cellula e cellula", que refleja el origen de las células existentes de otras células y describe la ontogenia como un proceso gradual de divisiones binarias. desde un óvulo fertilizado hasta tejidos adultos. La teoría celular clásica, por lo tanto, se basó en tres principios principales:
1. Todos los organismos vivos están compuestos por una o más células.
2. La célula es la unidad básica de estructura y función en todos los organismos.
3. Todas las células surgen de células preexistentes.
La historia de los descubrimientos que condujeron a una imagen unificada de la división celular (y la relación entre el núcleo y el citoplasma durante la formación de nuevas células) es un excelente ejemplo de cómo la elección de los métodos y el sistema modelo pueden influir en la teoría inferida. Este aspecto siempre ha limitado la biología experimental y sigue siendo relevante en nuestro tiempo. Desde el punto de vista contemporáneo (es decir, el juicio retrospectivo), las ideas sobre la formación extracelular y la cristalización alrededor de los núcleos pueden parecer oscuras. Sin embargo, hay que reconocer que muchas conclusiones se basaron en observaciones de tejidos fijos propensos a los artefactos y que solo proporcionan una visión estática de los fenómenos dinámicos subyacentes. La presencia de mitosis abierta tanto en animales como en plantas[40] hizo que descifrar las relaciones entre “savia” (citoplasma), núcleo y división celular fuera aún más complicado hasta que se entendió la naturaleza de los cromosomas. Además, algunos de los tejidos utilizados en el pasado como sistemas modelo se conocen hoy en día como casos bastante excepcionales. Incluso las observaciones originales de los tejidos vegetales por Schleiden involucraban el sincitio del endospermo sometido a celularización, lo que podría haberle dado una impresión errónea de la formación de células. Muchas de las primeras conclusiones también fueron engañadas al confundir granos de almidón (que se forman dentro de las células) con núcleos. Por otro lado, Dumortier y von Mohl pudieron hacer su descubrimiento sobresaliente mediante la observación de un sistema modelo ideal para el estudio de la división celular binaria: el alga filamentosa Conferva (Draparnaldia según la nomenclatura contemporánea) con células dividiéndose en los extremos de los filamentos. El cartílago se utilizó repetidamente como argumento del origen acelular de las células animales. Los embriones en desarrollo, que permitieron la observación directa de células en división no fijadas en el tiempo, fueron la fuente tanto de apoyo para un modelo de división celular binaria como de juicio erróneo. Aunque muchos autores (trabajando principalmente con modelos de anfibios) interpretaron correctamente la partición del huevo como una división celular progresiva, el biólogo francés Quatrefages afirmó a mediados del siglo XIX que el desarrollo de embriones de gasterópodos implica la formación de células dentro de las células. Quatrefages probablemente fue impulsado por un intento de apoyar el modelo de Schwann. Incluso Dumortier, quien descubrió la división binaria en Conferva (Draparnaldia por nomenclatura contemporánea), reconoció la posible formación de células dentro de las células e incluso la formación de células a partir de material acelular después de observar el desarrollo de los gasterópodos. Los embriones vitelinos grandes con división desigual también fueron fuente de confusión, como en el caso de Carl Vogt, quien afirmó que el surco del embrión de rana Alytes era independiente de la formación de nuevas células[41]. A la luz de observaciones fragmentarias incongruentes, fue honesto por parte de muchos científicos contemporáneos del siglo XIX reconocer la pluralidad de mecanismos de formación de células animales. Las afirmaciones universales sólidas requerían una comparación sistemática de muchos tejidos diferentes y técnicas mejoradas, como las realizó Remak. Aunque se opuso a las ideas que implicaban la formación intracelular de células, admitió que a menudo no era la negligencia en la observación o el mal juicio lo que conducía a conclusiones incorrectas, sino la elección accidental de material problemático como el cartílago o la fibra muscular. Sin embargo, incluso Remak llegó a una conclusión errónea con respecto a la división nuclear, posiblemente debido a la observación de muestras fijas estáticas y un sesgo hacia hacer una analogía entre la división celular binaria y la división nuclear binaria. Karl Bogislaus Reichert (1811-1883) observó la disolución de los núcleos durante la división de los glóbulos rojos, que utilizó como argumento para el concepto de Schwann de formación de núcleos de novo y contra el concepto de división celular binaria. Remak afirmó que no había podido reproducir la observación de Reichert de la disolución del núcleo en los glóbulos rojos en división. Las ideas de Reichert sobre la formación de células eran generalmente incorrectas, pero algunas de sus observaciones eran correctas, mientras que las ideas de Remak sobre la formación de células eran en general correctas, pero algunas de sus observaciones eran incorrectas. Remak ocasionalmente observó la disolución nuclear, pero la interpretó como un artefacto. Tanto Remak como Virchow apoyaron un modelo de división binaria nuclear que implicaba la formación de surcos, constricción y división de un núcleo en dos. Algunos científicos defendieron los modelos de Remak y Virchow, mientras que otros se refirieron a la disolución nuclear ("doctrina de Reichert"), a menudo con interpretaciones cercanas a las ideas originales de Schwann sobre la formación de células.
3.6 Conceptos protoplásmicos y críticas tempranas de la teoría celular recientemente establecida
La teoría celular fue popular entre los reduccionistas, quienes intentaron comprender los fenómenos fundamentales de la vida mediante el estudio de componentes estructurales simples. Las mejoras tecnológicas como la lente de inmersión en aceite, la técnica de micrótomo de Purkyne y los métodos novedosos de fijación y tinción[42] llevaron a innumerables observaciones de células y su contenido en el siglo XIX. También existieron críticas a la teoría celular y, en casos extremos, muchos descubrimientos histológicos fueron acusados de ser artefactos de tinción y / o fijación. El escepticismo sobre la universalidad de la teoría celular a menudo citaba la existencia de células sin núcleo, sincitios multinucleares y grandes cantidades de material extracelular en tejidos adultos como evidencia en contra de la teoría celular. Sin embargo, todos estos fenómenos se entendieron en última instancia como productos del desarrollo de las células. Uno de los últimos amargos argumentos sobre la validez general de la teoría celular fue sobre la naturaleza del tejido nervioso. La “teoría reticulada” consideraba el tejido nervioso como una red continua e ininterrumpida, debido a los límites de observación establecidos por los microscopios contemporáneos. Sin embargo, la teoría celular consideraba que el tejido nervioso estaba formado por células individuales como en otros tejidos (la "doctrina neuronal"). Ramón y Cajal demostró que esto último era cierto mediante el uso de un método de tinción que marcaba aleatoriamente solo unas pocas neuronas dentro del tejido, lo que indica claramente una discontinuidad en la red neuronal[43]. Además de las afirmaciones de que la teoría celular no puede explicar universalmente el funcionamiento de los organismos y que muchas estructuras observadas podrían ser artefactos de fijación, la teoría celular también fue acusada repetidamente de ser insuficiente o incluso no relevante para comprender las propiedades universales de la vida. Algunas de estas incongruencias se formularon en diversas formas de "teoría protoplásmica", que complementaron la teoría celular cerrando una brecha conceptual entre la superficie celular y el contenido celular o compitieron con la teoría celular cambiando completamente el enfoque de las células como simples ladrillos de construcción a la sustancia viva dentro de la célula. El término “protoplasma” fue introducido por Jan Evangelista, Purkinje en 1839, mucho antes que Hugo von Mohl y en un sentido muy similar. Hugo von Mohl criticó el enfoque de Schleiden y Schwann en la comprensión de las células en términos de límites y bloques de construcción y no le gustaban las analogías entre células y cristales. Redefinió la función de la célula como más basada en la organización interna y formuló su teoría protoplásmica en 1846[44]. Ferdinand Cohn propuso en 1850 que “las plantas y los animales eran análogos no solo por su construcción a partir de células, sino también, a un nivel más fundamental, en virtud de una sustancia común, el protoplasma, que llenaba las cavidades de esas células” (Liu 2016). Por lo tanto, conectó el concepto de von Mohl con la idea anterior de "sarcode", una sustancia contráctil propuesta por Fe'lix Dujardin para proporcionar la base de vida de los eucariotas unicelulares (Liu 2016). La tendencia a buscar atributos básicos de la vida (irritabilidad, sensibilidad, contractilidad, reproducción, etc.) en las propiedades del protoplasma no era infrecuente, y el protoplasma en sí se comparó con un "organismo elemental". El anatomista Max Schultze sugirió a mediados del siglo XIX que la verdadera base de la vida se encontraría estudiando el protoplasma, no la célula y redefinió la célula como un "grupo de protoplasma" alrededor de un núcleo (Liu 2016). Algunos autores consideraron la célula como una envoltura inerte y se centraron en estudiar el protoplasma como el "estado desnudo de la materia viva[45]". Por ejemplo, Wilson no afirmó que el protoplasma sea el único elemento vivo dentro de la célula: “El protoplasma privado de materia nuclear ha perdido, total o parcialmente, una de las propiedades vitales más características, a saber, el poder del metabolismo sintético, pero todavía hablamos de él como 'vivo', porque durante mucho tiempo puede realizar algunas de las otras funciones, manifestando irritabilidad y contractilidad, y mostrando también una coordinación definida de movimientos ”(como en el protozoo enucleado[46]). También hizo caso omiso de las versiones fuertes del reduccionismo que buscaban un solo elemento básico de la vida en "cualquier sustancia o elemento estructural de la célula", porque "la vida en su pleno sentido es propiedad del sistema celular como un todo más que de cualquiera de sus elementos separados ". Su teoría, por tanto, no es atomista ni reduccionista, sino que pone un fuerte énfasis en las propiedades del protoplasma al afirmar que “la sustancia continua es el elemento más constante y activo y que forma la base fundamental del sistema, transformándose en gránulos, gotas, fibrillas o redes de acuerdo con las diferentes necesidades fisiológicas”(Wilson 1899). Sin embargo, Wilson admitió proféticamente que no pudo llegar a ninguna conclusión general clara porque la base de todos los fenómenos radica en la "organización invisible de una sustancia que a simple vista parece homogénea". Creía que los "cuerpos ultramicroscópicos", moléculas, grupos de moléculas y micelas formaban la base de la organización protoplásmica (Wilson 1899).
3.7 Descubrimiento de orgánulos: mayor apreciación del contenido celular
Junto con los conceptos protoplásmicos que involucran las acciones de micelas, gotas y fibrillas diminutas, la presencia de estructuras más grandes localizadas dentro de las células fue reconocida y enfatizada cada vez más, incluida la noción de unidades vivas más pequeñas presentes. dentro de las células, inspirado en la teoría de la espontaneidad y jerarquía de mónadas de Leibnitz. Franz Unger describió las estructuras móviles en el citoplasma del polen como un “ejército de mónadas lleno de vitalidad interior, lleno de una autodeterminación interior que se revela en sus movimientos”. Las observaciones de eucariotas unicelulares grandes como las amebas y los ciliados estimularon aún más los pensamientos sobre estructuras subcelulares con funciones especializadas, análogas a los cuerpos macroscópicos. En 1884, Karl August Mobius sugirió el término "organulum" (pequeño órgano) para tales estructuras porque forman parte de una célula, mientras que los verdaderos órganos de los animales multicelulares consisten en muchas células. El término se transformó más tarde en "orgánulo" y su significado se amplió para cubrir las estructuras subcelulares de organismos unicelulares y multicelulares[47]. Van Benden y Boveri hicieron un avance importante a finales del siglo XIX. Descubrieron el ciclo de vida autónomo del centrosoma y concluyeron que la estructura tenía vida propia; Boveri describió el centrosoma como un órgano especial de división celular. Whitman percibió la célula como una "colonia de unidades más simples, núcleo, centrosoma, etc.", de la misma manera que un organismo superior es una colonia de células. En 1882, Julius Sachs escribió que los "cuerpos de clorofilo" (cloroplastos) se comportaban como organismos autónomos que se dividen para ajustar su número al tamaño de las hojas en crecimiento[48]. En 1883, Andreas Schimper notó la similitud entre los cloroplastos y las cianobacterias y propuso el origen cianobacteriano simbiótico de los plástidos. En 1890, Altmann postuló la presencia universal de "bioblastos" (llamado "Mitocondrias" por el microbiólogo alemán Benda en 1898) y descubrió que tenían las mismas propiedades de tinción que las bacterias; concluyó que eran bacterias modificadas. Esta idea del origen endosimbiótico de los cloroplastos y el origen xenobiótico de las células eucariotas como una amalgama evolutiva de organismos que alguna vez fueron independientes fue desarrollada por Konstantin Mereschkowsky entre 1905 y 1920[49], pero no fue generalmente aceptada hasta su fecha de lanzamiento. avivamiento en la década de 1970. Con microscopios y métodos de tinción mejorados, se agregaron orgánulos nuevos a los núcleos, cloroplastos y vacuolas conocidos de observaciones anteriores[50]. Con el descubrimiento del "ergatoplasma" (más tarde llamado "retículo endoplásmico") en 1897 y el aparato de Golgi un año después, la mayoría de los componentes comunes más grandes del "inventario" celular se conocían a fines del siglo XIX.
3.8 Disputas sobre los límites celulares
Para un sistema vivo, la existencia y las propiedades de un límite con el mundo exterior son tan importantes como las propiedades de su composición interna. Sin embargo, la presencia e identidad de un límite entre las células y el entorno exterior no estaba clara en el siglo XIX y (especialmente desde la perspectiva contemporánea) fue ignorada en gran medida por los defensores de los puntos de vista tanto celular como protoplásmico. Schwann asumió que las superficies/membranas siempre limitan la movilidad dentro / fuera de una célula, incluso si es invisible, y esto podría inferirse del movimiento browniano de los componentes celulares, que no escapan del volumen celular delimitado por la estructura de la superficie. Sin embargo, en general, la comparación de las superficies celulares de las células vegetales (con paredes) y las células animales era confusa y los términos "pared" y "membrana" se usaban indistintamente. Las membranas verdaderas eran imposibles de detectar con las técnicas de histología del siglo XIX. Así, en la segunda mitad del siglo XIX, se prestó poca atención a las membranas y, si estaban presentes, se las consideró estructuras secundarias no esenciales originadas por el endurecimiento de la superficie celular. Max Schulze, el proponente de la teoría protoplásmica, también fue un entusiasta oponente del concepto de membrana[51]. Postuló, en lugar de células, pequeñas ampollas de protoplasma contráctil inmiscible con agua. Las membranas detectadas fueron simplemente el resultado del endurecimiento del protoplasma causado por el contacto con el ambiente exterior o un artefacto de degeneración y el sello distintivo del material celular muerto. El principal apoyo para el concepto de membrana provino de estudios osmóticos. Hewson publicó experimentos sobre la hinchazón y el encogimiento de las células sanguíneas ya en 1773. En la primera mitad del siglo XIX, Dutrochet explicó la turgencia de las plantas por ósmosis a través de un borde con “tamices químicos”. Las primeras membranas artificiales se crearon por precipitación de ferrocianuro de cobre (a partir de ferrocianuro de potasio y sulfato de cobre) y, por lo tanto, se denominaron membranas de precipitación. Junto con el concepto coloidal contemporáneo de los interiores celulares y las ideas sobre las membranas celulares que se originan a través del endurecimiento de la superficie, la existencia de membranas de precipitación artificial alimentó la creencia de que la superficie del protoplasma coloidal se precipita y forma una barrera osmótica. Los experimentos pioneros de Overton (publicados entre 1895 y 1900) mostraron cambios en el volumen celular en más de 500 soluciones diferentes y le permitieron concluir que debe existir una barrera distinta de la pared celular de la planta y está hecha de componentes solubles en éter (es decir, es hidrofóbica). Sugirió colesterol y fosfolípidos como posibles candidatos. En combinación con trabajos sobre electrofisiología y experimentos de microinyección, la aceptación de la membrana plasmática como una estructura real se estableció a principios del siglo XX.
3.9 Hacia los determinantes celulares de la herencia
Una imagen clara de la división nuclear se formó solo después de que se descubrieron y comprendieron el huso mitótico y los cromosomas. Las observaciones recurrentes finalmente llevaron al consenso de que los núcleos se desmontan y se vuelven a montar durante la división celular. Strassburger propuso la homología de la división de células vegetales y animales antes de finales del siglo XIX. En la década de 1870, los detalles de los eventos de división celular se observaron repetidamente y, en 1879, Walter Flemming acuñó el término "proceso mitótico" y describió su cronología básica. Flemming también introdujo el término "cromatina" y fue el primero en describir la división longitudinal de los cromosomas en células tanto animales como vegetales. Fue un crítico agudo del concepto de división nuclear directa defendido por Remak y Virchow, pero al mismo tiempo reconoció plenamente la continuidad del material nuclear durante la división celular al expandir la declaración de Virchow en "Omnis nucleus e nucleo". En ese momento, también hubo un gran esfuerzo por localizar los determinantes materiales de la herencia. Muchos grandes biólogos del siglo XIX, aunque no trabajaran con las células mismas, postularon tales partículas (Darwin postuló gemulae; Haeckel, plastiduls; Spencer, unidades fisiológicas; de Vries, pangenes; Galton, tiras, etc.) y así estimularon la búsqueda. para ellos (Radl 1930). El trabajo descriptivo acumulativo ayudó a caracterizar la progresión de la división celular y el comportamiento de los cromosomas con suficiente detalle como para que fueran posibles las interpretaciones biológicas y los experimentos de manipulación. Ya en 1885, A. Weissmann propuso el concepto de bucles cromosómicos como lugar de almacenamiento de información hereditaria y ayudó a explicar los fenómenos de meiosis y recombinación. El trabajo de Theodor Boveri (1862-1915) no solo demostró definitivamente la función cromosómica en la herencia, sino que también cambió el trabajo de una combinación de observaciones y deducción a la introducción de experimentos de manipulación. Sus experimentos con embriones de erizo de mar involucraron polispermia y manipulación de la escisión temprana del embrión, lo que resultó en blastómeros con una distribución cromosómica desigual. Boveri descubrió que el destino de los blastómeros se correlacionó con las anomalías cromosómicas introducidas y dedujo que los diferentes cromosomas tienen diferentes cargas genéticas. Después del redescubrimiento de según las leyes de Mendel, Boveri fue el primero en señalar la similitud entre la segregación de elementos, según lo propuesto por Mendel, y la segregación física de los cromosomas. El primer concepto de genes era puramente fenomenológico y no necesariamente preguntaba por el agente material de la herencia. Posteriormente, a la luz de las tendencias mecanicistas, se concibió un componente material responsable de la transmisión de información genética. Boveri propuso que la base material de las leyes de herencia de Mendel radicaba en las propiedades de los cromosomas y, por lo tanto, contribuyó al desarrollo de la genética molecular en el siglo XX.
3.10 Células en los tejidos: primeros enfoques experimentales holísticos y reduccionistas
Desde los primeros días de la teoría celular, muchos científicos han subrayado que los organismos son más que un simple ensamblaje de sus partes, y que los aspectos funcionales de la vida deben estudiarse en el contexto del conjunto. embrión / organismo en desarrollo. Las actitudes iban desde la crítica aguda de la doctrina celular como insuficiente y engañosa, pasando por los intentos de introducir principios organizativos novedosos que complementaran y coordinaran la acción de las células, hasta un intento sistemático de comprender los embriones en desarrollo puramente a partir de las interacciones colectivas de las células individuales. T.H. Huxley propuso una interpretación fisiológica de la célula en oposición al concepto morfológico de Schleiden y Schwann. Afirmó que "la teoría celular de Schleiden y Schwann" no solo "se basaba en concepciones erróneas de la estructura", sino que también conducía "a errores de fisiología[52]". En particular, le disgustaba que la "doctrina celular" exagerara el supuesto de la individualidad anatómica de las células y consideró que las células deberían estudiarse en su relación mutua en el contexto del desarrollo, porque toda la historia de vida de un organismo está "dominada por el desarrollo". Whitman afirmó que “el hecho de que la unidad fisiológica no se rompa por los límites celulares se confirma de tantas maneras que debe aceptarse como una de las verdades fundamentales en biología”. Sachs defendía el punto de vista del organismo y consideraba que la presencia de células, aunque era un fenómeno general de la vida, era de importancia secundaria y sólo una de las muchas manifestaciones de las fuerzas formativas de la vida. La idea de Sachs de que el crecimiento y el cambio de las formas de las plantas es primario y que los planos de división celular son secundarios y dependen del crecimiento general también fue compartida por de Bary, quien acuñó la famosa afirmación: “La planta forma células, el las células no forman plantas ”(Thompson 1917). Los principales intentos de análisis causal del desarrollo embrionario como resultado de la interacción colectiva de células individuales cristalizaron en la disciplina de Entwicklungsmechanik (mecánica del desarrollo en el sentido de causalidad natural), defendida con entusiasmo por Wilhelm Roux. Roux cambió el enfoque de las especulaciones basadas puramente en observaciones descriptivas a experimentos manipuladores en una búsqueda de una explicación causal del desarrollo por combinación de fuerzas de actuación individuales[53]. Basado en sus experimentos con embriones de anfibios, Roux abogó por un concepto de mosaico de desarrollo, afirmando que las células del embrión temprano determinan la posición de las partes posteriores del organismo. Otros científicos propusieron diferentes conceptos de desarrollo, en gran parte porque utilizaron otros sistemas modelo, como los cnidarios y los embriones en desarrollo temprano que muestran una asombrosa capacidad de regeneración y un cierto grado de invariancia de morfogénesis con respecto al número de células participantes. Tales experimentos sugirieron que las células del mismo linaje pueden tener diferentes destinos y las células de diferentes linajes el mismo destino, dependiendo de la posición que adquieran dentro del embrión. Whitman afirmó que “la embriología comparada nos recuerda a cada paso que el organismo domina la formación celular, utilizando para el mismo propósito una, varias o muchas células, acumulando su material y dirigiendo sus movimientos, y dando forma a sus órganos, como si las células no lo hicieran. existen, o como si existieran sólo en completa subordinación a su voluntad ”.
Algunas de las tendencias incluso dieron como resultado la búsqueda de principios holísticos que preceden a la formación de células y organizan las acciones de las células en todo el organismo en desarrollo. Hans Driesch también intentó romper el proceso continuo de morfogénesis animal en sus elementos últimos (primeros principios) al comienzo de su carrera[54]. De una manera visionaria, consideró que el desarrollo "comenzaría con unas pocas variedades ordenadas", que gradualmente "crearían, mediante interacciones, nuevas variedades", que "actuando sobre las originales (variedades) provocan nuevas diferencias". "Con cada respuesta, se proporciona de inmediato una nueva causa y una nueva reactividad específica para otras respuestas específicas". Partes del embrión en desarrollo constituyen así una conversión gradual de estados y receptividad a otros estímulos. Gobernado por el núcleo, los químicos organogenéticos se forman en el citoplasma, que actúa como intermediarios entre los estímulos externos y el núcleo. Una cascada de estímulos entre las células y sus activaciones parciales impulsan el desarrollo del organismo. Más adelante en su vida, Driesch se volvió crítico de sobrestimar la teoría celular potencial explicativa (Whitman 1893) e incluso revocó algunas de sus posiciones originales. Experimentos con cnidarios, mohos de limo acrasido, plantas y embriones de equinodermo lo llevaron a buscar leyes fundamentales que determinan el sistema de coordinación espacio-temporal que lleva a las células a la forma. Driesch defendió un enfoque matemático y físico pero también quiso que la biología fuera una ciencia con autonomía y, por lo tanto, buscó principios de organización en torno a los cuales se constituyan los fenómenos químicos y físicos en proceso. Su conclusión de que la química y la física contemporáneas no eran suficientes para explicar la embriogénesis podría, de hecho, extenderse hasta la década de 1970, cuando la investigación celular incorporó avances en cibernética y genética. Driesch puso un fuerte énfasis en la teleología en el desarrollo y trató sin éxito de formular la entelequia como una nueva cantidad física colectiva, específico para organismos, que podría analizarse mediante enfoques matemáticos.
El intento de Driesch de descubrir las leyes de organización típicas de la biología fue desarrollado por Alexander Gurwitsch[55]. Gurwitsch estudió el desarrollo del cerebro de tiburón, los cuerpos fructíferos de los hongos y las flores compuestas y llegó a la conclusión general de que la forma general se desarrolla repetidamente de manera exacta a pesar de las fluctuaciones en la forma y la tasa de crecimiento de las partes individuales. También pensó que el contorno de una parte o de un embrión completo se puede formular matemáticamente con más precisión que la forma y disposición de sus componentes internos. Buscando un principio supracelular que ordene y coordine las células sobre el embrión, e inspirado en los desarrollos contemporáneos de la física, formuló el concepto de un “campo específico de la especie” que organiza la morfogénesis[56]. Las células producen el campo que se extiende y afecta a un espacio extracelular y, al mismo tiempo, el campo actúa sobre las células. Los campos de las células forman un campo agregado, que depende de la configuración del conjunto multicelular y existe una retroalimentación entre el campo y sus consecuencias morfogenéticas. La interdependencia entre las propiedades de las células y sus coordenadas de posición dentro de un organismo en desarrollo debe ser precisa y matemáticamente simple. Gurwitsch incluso intentó definir el campo de manera vectorial (como una descripción geométrica, no en un sentido estrictamente físico), donde las células seguían los vectores del campo. En la década de 1930, se habían logrado muchos descubrimientos cruciales en embriología experimental. Muchos estudios incluyeron el aislamiento y la recombinación de partes embrionarias y el mapeo de la diferenciación y el potencial inductivo de las partes aisladas de los embriones y los efectos de las partes trasplantadas a otros embriones, incluidos los trasplantes interespecíficos[57]. Se estudiaron intensamente fenómenos como el potencial inductivo de los pliegues neurales y el establecimiento de la polaridad de las extremidades. Hans Spemann reintrodujo el término "campo de organización" para describir las propiedades inductivas del blastoporo dorsal anfibio[58], basándose conceptualmente en el concepto de Driesch de un "sistema equipotencial armonioso". Por tanto, el concepto de campo seguía siendo vital y, en 1939, Paul Weiss postuló que el campo es el principio organizador clave de la embriología; los fenómenos del desarrollo tienen propiedades de campo y los componentes de los campos están conectados por una red de interacciones. Los conceptos de campo en la embriología experimental de la década de 1930 eran materialistas. Weiss afirmó que el campo tiene existencia física y está limitado por sustratos físicos de los que surge la morfogénesis y debería ser objeto de investigación como cualquier otro fenómeno físico. Se suponía que el campo morfogenético se convertiría en el paradigma básico de la embriología en su intento de descubrir las leyes de la morfogénesis.
3.11 Establecimiento de la biología molecular
Los detalles del nacimiento y la historia temprana de la bioquímica están más allá del alcance de esta revisión. Sin embargo, mencionamos varios descubrimientos y conceptos clave porque el paradigma y la metodología elaborados por los bioquímicos influyeron en gran medida en el advenimiento de la biología celular moderna, especialmente en el siglo XX. Aunque la mayoría de los científicos alemanes que estudian las células se centraron en su estructura y formación, el naturalista francés Francois Vincent Raspail (1794-1878) estaba interesado en la química de las células[59]. Analizó la composición química de las células mediante la adopción de análisis de combustión química para muestras pequeñas (microcombustión) y desarrolló procedimientos de tinción para detectar almidón, albúmina, sílice, mucina, azúcar, cloruros y hierro. También enfatizó que la célula es en sí misma un microlaboratorio que equilibra cuidadosamente el catabolismo y el anabolismo. En 1833, Payen y Persoz purificaron una fracción termolábil capaz de descomponer el almidón en azúcar. Estos "agentes" fueron posteriormente denominados enzimas por Wilhelm Kuhne. En 1893, Eduard Buchner pudo replicar todo el proceso de fermentación de la levadura mediante un extracto sin células. Thomas Burr Osborne cristalizó proteínas sistemáticamente y demostró una gran diversidad de especies de proteínas[60]. En 1926, James Sumner logró aislar y cristalizar una enzima (ureasa) por primera vez. Redisolvió la ureasa del cristal (por lo tanto, libre de cualquier pequeño compuesto potencialmente co-purificado de la célula) y mostró su actividad catalítica, demostrando también la naturaleza proteica (y biopolimérica) de las enzimas[61]. El enfoque inicial de la bioquímica fue, por tanto, ortogonal al de la microscopía. Las propiedades de la vida se estudiarían fuera del contexto del organismo, independientemente de los principios estructurales del cuerpo intacto. El objetivo era replicar la vida o procesos similares a la vida en un sistema aislado con un conjunto mínimo de componentes y, por lo tanto, aislar las sustancias subyacentes para comprender las propiedades y los cambios en curso de la materia. Partes del concepto "protoplásmico" fueron abandonados o eclipsados ??por el advenimiento de la bioquímica clásica, que se centró en moléculas aisladas en soluciones de agua tamponada de composición simple. La percepción simplificada de "bolsa de enzimas en solución" del contenido celular, donde las moléculas se encuentran al azar y siguen la ley de acción de masas, fue criticada al comienzo de la ciencia de la bioquímica. Se sugirió que los agentes catalíticos actúan como parte de una red proteica integral y dinámica en la célula. Sin embargo, el enfoque original de la bioquímica temprana en las enzimas como agentes catalíticos proporcionó un conjunto de herramientas mecanicistas unificadas para caracterizar subconjuntos de componentes y fenómenos celulares. Actualmente se entiende que la biología molecular se basa en la genética molecular, pero antes de que se adquiriera la capacidad de modificar la información genética, fue la bioquímica la que estableció la primera verdadera descripción reduccionista a nivel molecular de algunos procesos de la vida. La síntesis de las teorías mendeliana y de la herencia cromosómica a principios del siglo XX colocó a los genes en el contexto espacial de ubicación en los cromosomas y estimuló la institucionalización de la genética como disciplina. Como resultado de la con un enfoque reduccionista exitoso y el impacto económico inmediato en la reproducción, hubo una tendencia común a poner la genética en el centro de un marco de biología mecanicista[62]. Por ejemplo, la genética del desarrollo surgió como un programa alternativo que competía con la embriología experimental establecida (en lugar de proponerse como un enfoque complementario). Tanto el concepto de gen utilizado por los genetistas como el concepto de campo utilizado por los embriólogos eran abstractos y se consideraba que ambos tenían una base física, aunque comprendidos sólo de forma vaga. En ese momento, todavía se consideraba que los genes estaban asociados con la acción de proteínas, posiblemente enzimas. Las tendencias genocéntricas fueron así evidentes en biología al menos dos décadas antes de que se consolidaran los principios de la biología molecular. El concepto de campo como principio organizador fue finalmente abandonado, en gran parte porque no se disponía de técnicas bioquímicas para examinar los fenómenos de campo en detalle, mientras que las técnicas para el estudio de la expresión génica en sistemas modelo aparecieron gradualmente. A pesar de los continuos intentos de interpretar la vida en un marco o perspectiva holística, los enfoques reduccionistas prevalecieron en biología como un marco pragmático para encontrar explicaciones mecanicistas de fenómenos complejos. La genética, la bioquímica y la biofísica se desarrollaron de forma independiente durante algún tiempo, pero comenzaron a converger después de la década de 1930. Experimentos clave sobre la regulación genética de la bioquímica de Neurospora en la década de 1940 mostraron que cada paso en una vía metabólica está controlado por un solo gen y esto llevó a la "hipótesis de un gen, una enzima", que sugirió que cada gen actúa directamente como una enzima o determina la especificidad de una enzima. Esto estimuló aún más la percepción del gen como una unidad central de la función biológica y gran parte de la atención se centró en la relación entre el ácido nucleico y las macromoléculas de proteínas y la búsqueda de la base molecular de la herencia. La introducción de técnicas novedosas como la cristalografía de rayos X y la ultracentrifugación ayudó a cambiar el enfoque de las teorías coloidales a los biopolímeros y sus estructuras. Recapitular los grandes esfuerzos de la biología molecular del siglo XX está más allá del alcance de esta revisión y se describe detalladamente en otra parte. Lo más importante es que se descubrió la base material de la información hereditaria en forma de secuencias de nucleótidos de ácidos nucleicos y se resolvió el código genético, descubriendo la relación entre una secuencia de genes y la macromolécula de proteína que codifica. Los descubrimientos de los principios básicos de la biología molecular estimularon aún más la búsqueda de genes responsables de todo tipo de procesos en los organismos vivos. Con las rutas metabólicas básicas mapeadas, los bioquímicos se interesaron en la regulación del metabolismo. Después de la investigación pionera de Jacques Monod (1910-1974) sobre la regulación de las vías bioquímicas y la expresión génica[63], los conceptos de retroalimentación positiva, retroalimentación negativa, regulación alostérica, cooperatividad, inducción de enzimas, control por represión, La regulación no lineal, la inhibición cruzada y la integración booleana de los procesos reguladores se convirtieron en el vocabulario estándar de la biología molecular[64]. Los paralelos entre la biología molecular y la cibernética se basaron así (Monod 1972), aunque las ideas sobre la señalización celular y la expresión génica en la época tenía sus raíces en la bioquímica y las simples relaciones cibernéticas. Las herramientas recientemente desarrolladas cambiaron el enfoque hacia el estudio de genes individuales y sus productos proteicos o rutas simples de señalización, genética y bioquímica. Se entendió que otros componentes, como los componentes de la matriz extracelular (MEC) y la composición de lípidos de la membrana, también desempeñan funciones importantes (Monod 1972) pero, debido a dificultades tecnológicas, se descuidaron en comparación con la investigación realizada sobre el ADN y las proteínas. Se entendió que estas moléculas estaban localizadas dentro de las células, pero se puso más énfasis en comprender su función a nivel molecular que en sus funciones celulares en términos de organización estructural de las células.
3.12 Membranas biológicas en el siglo XX: del descubrimiento de las bicapas lipídicas al modelo de mosaico fluido
A pesar del descuido inicial de la barrera celular en el siglo XIX, la naturaleza de las membranas biológicas se convirtió en un tema importante en la biología celular del siglo XX. En 1925, Gorter y Grendel realizaron un experimento pionero que abordaba la naturaleza estructural de la membrana plasmática. Escogieron eritrocitos, células desprovistas de membranas internas, como sistema modelo y mostraron que la proporción de área de monocapa formada a partir de lípidos extraídos y área de superficie de eritrocitos era 2: 1, lo que sugiere la naturaleza bicapa de la membrana plasmática[65]. Cabe señalar que el experimento fue criticado por varias deficiencias, incluido el descuido de los componentes proteicos de la membrana plasmática y el cálculo incorrecto de la superficie de los eritrocitos. Ahora se cree que varios errores experimentales se cancelaron recíprocamente entre sí, lo que lleva a la conclusión correcta. Sin embargo, la validez de este primer modelo solo puede apreciarse a la luz de experimentos mucho más tardíos. Independientemente de las críticas, el impacto inmediato de la hipótesis de la bicapa lipídica fue abrir una discusión sobre la naturaleza molecular de la estructura de la membrana. Las tendencias basadas en las membranas de precipitación de Traube y las membranas lipídicas de Overton fueron populares. En términos de predicción de la permeabilidad de las moléculas, un componente crucial del primero fue el tamaño de los poros y del segundo, la hidrofobicidad. Las teorías unificadoras asumieron que las membranas eran capas de lípidos interrumpidas por poros. Se reconocieron las funciones mixtas de los lípidos y las proteínas en la función de las membranas, pero su contribución relativa fue un tema controvertido (Lombard 2017). Además del carácter icónico de la búsqueda de determinantes de la herencia molecular y la resolución del papel diferencial de las proteínas y los ácidos nucleicos en el núcleo, otra cuestión clave en la biología celular del siglo XX fue la naturaleza de la interacción de proteínas y lípidos en el funcionamiento de las membranas biológicas. Varios modelos involucraron mezclas de fracciones de lípidos y proteínas dentro o entre capas postuladas de la membrana. Curiosamente, uno de los conceptos que dominó la investigación de membranas durante décadas fue el "modelo paucimolecular", que postulaba una capa de lípidos intercalada entre dos capas de proteínas. El modelo se basó en la medición de la tensión superficial entre las células de equinodermo/teloostei y una capa de aceite, así como la estructura de axones mielinizados. Los experimentos de tensión superficial pronto fueron criticados por usar triacilglicerol en lugar de fosfolípidos de membrana nativos y por usar axones mielinizados como modelo representativo de una membrana celular general. Sin embargo, el concepto se hizo popular durante mucho tiempo y se interpretó que las primeras Imag3 de microscopía electrónica (EM) de baja calidad respaldaban el modelo de membrana paucimolecular. Como en muchos otros casos, un conjunto de experimentos bien intencionados y la elección del sistema modelo llevaron a suposiciones erróneas que persistieron durante décadas (Lombard 2014). Los modelos en mosaico de la membrana plasmática también fueron populares. Las especulaciones que involucran partes similares a grasas y partes similares a protoplásmicas, una mezcla de elementos solventes y similares a tamices, fueron respaldadas por experimentos de permeabilidad a principios del siglo XX. Los experimentos de permeabilidad también sugirieron que el diámetro del "poro" podría cambiar de acuerdo con la hidratación del poro, el pH, la actividad metabólica y el tipo de célula, pero los mecanismos moleculares de las propiedades de la membrana no estaban claros. Incluso los experimentos innovadores de Hodgkin y Huxley sobre la excitabilidad de la membrana (1952) fueron fenomenológicos y no se conocía el mecanismo de la permeabilidad diferencial de la membrana hacia los iones Na + y K + (Lombard 2017). Debido a que los iones de Na + hidratados son más grandes que los iones de K + hidratados, los agentes proteicos selectivos que facilitan el transporte de Na + eran difíciles de imaginar. Se postularon portadores basados ??en lípidos específicos para Na +. Además, varios argumentos en contra de la naturaleza lipídica de las membranas plasmáticas se basaron en su alta permeabilidad al agua. Estos enigmas se resolvieron finalmente en el contexto de una estructura delicada del canal de potasio y el descubrimiento tardío de las acuaporinas, proteínas de membrana que facilitan la permeabilidad del agua. El modelo de mosaico fluido dominó el campo de las membranas en la década de 1970. Era compatible con la mayoría de los experimentos contemporáneos y predecía observaciones futuras; el modelo permaneció básicamente inalterado durante las próximas décadas. Una de sus principales ventajas sobre varios modelos competidores fue la compatibilidad con la termodinámica de la unión proteína-lípido y lípido-lípido dentro de las membranas, en gran parte basada en interacciones hidrofóbicas[66]. El enfoque general en las proteínas fue fomentado por herramientas desarrolladas para la biología molecular, lo que resultó en que las proteínas de membrana fueran el objetivo principal de la investigación en busca de agentes moleculares con funciones de membrana particulares. Se consideró que los lípidos eran elementos estructurales pasivos que aseguraban principalmente la fluidez de las proteínas dentro de la membrana. Esta idea todavía se defiende en muchos libros de texto.
3.13 Perspectivas sobre la ultraestructura celular y el origen de los orgánulos en el siglo XX
El esfuerzo descriptivo clásico de la teoría celular continuó durante el siglo XX con las disciplinas de histología y citología. La barrera metodológica de la microscopía se rompió en la década de 1930 con la introducción de la microscopía electrónica.
En combinación con técnicas novedosas de fijación, corte y tinción, fue posible obtener Imag3 de estructuras subcelulares con una precisión de decenas de nanómetros. Las primeras Imag3 EM de las mitocondrias revelaron inmediatamente la presencia de una doble membrana con pliegues de la membrana interna, denominada crestas[67]. En 1953, EM ayudó a redescubrir el retículo endoplásmico[68]. La EM no solo sirvió como una herramienta para descubrir detalles novedosos de las estructuras subcelulares, sino que también aportó una confirmación independiente de las conclusiones sobre algunos acertijos o preguntas más antiguos. Por ejemplo, existían varios modelos en competencia de la estructura de la membrana plasmática y Fischer todavía se oponía a la teoría de la membrana en 1921, argumentando que las membranas eran invisibles incluso cuando los límites de las células eran visibles. La EM finalmente confirmó la presencia de una bicapa lipídica de la membrana plasmática incluso en las células bacterianas, donde su presencia se había debatido durante mucho tiempo (Lombard 2017). La teoría neuronal generalmente aceptada también se confirmó inequívocamente al visualizar la hendidura sináptica, un pequeño espacio entre las células nerviosas vecinas. La alta resolución espacial permitió la detección de nuevas estructuras de ramificación fina que conectan otros componentes celulares[69]. Esta red microtrabecular fue considerada el "componente sólido básico del citoplasma", pero también fue considerada un artefacto de fijación por muchos oponentes. El concepto de fases sólidas/líquidas y la heterogeneidad del citoplasma se convirtió en un tema candente durante algún tiempo, pero luego desapareció, solo para volver en los últimos años. La idea de simbiogénesis (introducida por Mereschkowsky) como la aparición de novedades evolutivas, incluidos los nuevos orgánulos celulares, fue revivida por Lynn Margulis en la década de 1970[70]. Margulis también propagó el concepto de endosimbiosis en serie, afirmando que las células eucariotas modernas se originaron por múltiples eventos simbiogenéticos sucesivos de organismos una vez independientes, y la idea de que los eventos simbiogenéticos eran una fuerza impulsora común en la especiación eucariota[71]. Con el empleo de técnicas de biología molecular, pronto se acumuló el apoyo al origen endosimbiótico de las mitocondrias y plastidios y el paradigma de la evolución de las células eucariotas pasó de la acumulación gradual de cambios como único mecanismo a la posibilidad de una adquisición abrupta de orgánulos. El resurgimiento del concepto de orgánulo simbiogenético y la idea de la célula eucariota como producto de la fusión celular entre Archea y Eubacteria[72] apunta al papel crucial de los procesos cooperativos en la evolución de la vida y al hecho de que la evolución de las células no podían entenderse completamente como un simple proceso progresivo e incremental, sino que involucraban singularidades con un impacto macroevolutivo crucial.
3.14 Formación de la biología celular moderna y tendencias metodológicas en la biología celular del siglo XXI
Mientras que la biología del siglo XIX tenía que decidir cuáles de las grandes teorías eran correctas, la biología celular de finales del siglo XX estuvo marcada por la tendencia a unir los descubrimientos de la genética, biología, bioquímica y citología en un todo congruente. Los enfoques de arriba hacia abajo (observación cada vez más detallada de la ultraestructura del tejido) y de abajo hacia arriba (examinando las propiedades de los componentes funcionales más pequeños en forma de moléculas y sus relaciones) se utilizaron finalmente juntos como un conjunto de herramientas comunes de un campo científico unificado. Muchos procesos se atribuyeron a genes específicos y sus productos proteicos. Las proteínas se mapearon con éxito en rutas bioquímicas, de señalización y reguladoras de genes. Con la ayuda de técnicas de fraccionamiento celular y EM, combinados con tinción de anticuerpos, fue posible mapear rutas bioquímicas y actividades de proteínas en compartimentos subcelulares específicos[73]. La capacidad de mantener, hacer crecer y manipular células fuera de los organismos (una tarea relativamente simple para las células vegetales), junto con la expansión de las técnicas de Imag3 de células vivas, especialmente el descubrimiento de proteínas fluorescentes codificadas genéticamente, llevó a innumerables observaciones de procesos dinámicos en células vivas. Las células siempre se han percibido como entidades dinámicas, pero las nuevas técnicas permitieron la observación de procesos moleculares in vivo con el contexto espacial y temporal adecuado. El énfasis se ha desplazado gradualmente del papel de los genes individuales a cómo las acciones de los componentes individuales dentro de la célula contribuyen colectivamente a un proceso particular. Esta tendencia no niega los descubrimientos anteriores de la biología molecular del siglo XX en ningún sentido, pero demuestra la importancia de estudiar los componentes moleculares dentro de las células vivas, teniendo en cuenta las propiedades estructurales y dinámicas del entorno celular. Así, la célula ha resurgido como una plataforma tanto biológica como interpretativa, que conecta los mecanismos moleculares con los fenómenos macroscópicos. Varias tendencias tecnológicas son típicas de la biología celular en este nuevo milenio. En primer lugar, las técnicas mejoradas permiten ahora seguir los procesos y componentes celulares con mayor y mayor precisión. La resolución de los microscopios de fluorescencia está aumentando en el tiempo y el espacio, más allá de la limitación impuesta por la barrera de difracción[74]. El límite de resolución clásico de la microscopía óptica se ha superado mediante la combinación de tecnologías de fluorescencia y métodos especializados de excitación de fluoróforos. Estas técnicas, junto con análisis informáticos sofisticados, permiten una resolución casi angstrom (Å) en casos específicos[75]. Los análisis estructurales de grandes máquinas macromoleculares como el ribosoma[76] y el poro nuclear[77] no son infrecuentes. Herramientas rápidas para la manipulación intracelular, como pinzas ópticas[78], proteínas activadas optogenéticamente y pequeñas moléculas fotoactivadas[79], ahora complementan las herramientas genéticas y farmacológicas tradicionales.
Algunas de las nuevas técnicas están ayudando a tender un puente sobre los enfoques tradicionales. Por ejemplo, la microscopía correlativa de luz y electrónica permite obtener Imag3 de células vivas. Se pueden adquirir datos EM de alta resolución para una parte específica de la celda después de la congelación rápida de la muestra en un momento determinado[80]. Durante la espectrometría de masas de Imag3, las regiones específicas de una célula/tejido se analizan por separado mediante espectrometría de masas, que por lo tanto se enriquece con información espacial[81]. En algunos casos, los análisis de las propiedades estructurales de las proteínas, obtenidos previamente mediante mediciones in vitro, se pueden realizar dentro del entorno celular[82]. Otra tendencia dominante de la biología celular contemporánea es el aumento del rendimiento experimental con la ayuda de la adquisición y el procesamiento de datos automatizados. Estas tendencias se introdujeron en gran medida para la secuenciación de genomas y transcriptomas completos, pero los enfoques "ómicos" se están generalizando en relación con la mayoría de las técnicas, incluida la microscopía de fluorescencia[83], la clasificación celular[84], la microscopía electrónica[85] y biología estructural[86].
3.15 Biología celular modular
Se ha hecho evidente que, aunque algunas funciones celulares simples son ejecutadas por un solo componente molecular (transporte de potasio a través de la membrana plasmática a través de un canal de membrana, conversión de metabolitos por una enzima especializada), la mayoría de las funciones celulares (regulación del crecimiento, diferenciación, quimiotaxis) surgen de las interacciones de muchos componentes[87]. Después de décadas de caracterizar los componentes y las tendencias de las células individuales para su catalogación total, el enfoque ahora está cambiando de identificar partes individuales a comprender sus relaciones, asociaciones espacio-temporales y comportamiento colectivo. Los enfoques de biología de sistemas se basan en la combinación de datos de alto rendimiento generados por varios análisis ómicos y computacionales cuantitativos para generar nuevos conocimientos integrados sobre cómo las partes individuales producen fenómenos emergentes. La definición y metodología precisas de la biología de sistemas no está unificada y, a menudo, es difícil de alcanzar[88], pero el énfasis principal está en deducir las propiedades de las redes de interacción que gobiernan los procesos celulares. Existe un debate en curso sobre la necesidad de cambiar la percepción y el lenguaje científico si queremos comprender las funciones celulares. El concepto de "biología modular" (estrechamente vinculado con el concepto de biología sintética) se basa en la comprensión de que los enfoques ómicos por sí solos no pueden descubrir y comprender los principios de "diseño" o "ingeniería" (en un sentido funcional) de los organismos vivos. Yuri Lazebnik ha pedido un nuevo lenguaje formalizado que sea más adecuado para comprender módulos en sistemas vivos[89]. Por otro lado, el lenguaje formal de la electrónica (con componentes como disparadores y amplificadores) que utilizan los ingenieros proporciona una visión directa de los procesos para los que los componentes están conectados. La analogía no es del todo justa porque los ingenieros han diseñado artefactos a partir de los primeros principios y han formulado un lenguaje adecuado en el camino, mientras que el enfoque de ingeniería inversa de la biología molecular se encuentra con sistemas que han evolucionado por sí mismos durante miles de millones de años en entornos complejos. Sin embargo, los biólogos podrían aprender más adoptando una perspectiva de ingeniería. Incluso los modelos originales de regulación de la expresión génica se inspiraron en la lógica booleana, y muchas máquinas modernas son ahora lo suficientemente complejas como para fomentar un mayor diálogo entre la biología y la ingeniería, al menos en el ámbito de la transmisión, el procesamiento y la interpretación de señales[90]. Los conceptos de amplificación, adaptación (a corto y largo plazo), robustez, aislamiento, atractores, biestabilidad, ondas y oscilaciones, interruptores de memoria, filtrado, reconocimiento de patrones, discriminación de series de tiempo, histéresis, operaciones de compuertas lógicas complejas, corrección de errores y coincidencia. la detección debería convertirse en una parte básica del vocabulario de biología celular. Los módulos celulares que reflejan estos conceptos, en lugar de moléculas individuales, son de interés principal para comprender los fenómenos celulares colectivos[91]. Se pueden utilizar métodos bioinformáticos novedosos para buscar motivos de red similares, y se puede probar experimentalmente si motivos similares desempeñan el mismo papel en diferentes contextos. Las funciones generales de retroalimentación positiva (biestabilidad, memoria, comportamiento similar a un interruptor) y retroalimentación negativa (resistencia al ruido, estado estable inducido por entrada) se conocen desde hace mucho tiempo. La lista de motivos y arquitecturas comunes asociados con funciones específicas en las células se está ampliando ahora. Por ejemplo, los bucles de retroalimentación coherentes a menudo actúan como detectores de persistencia, que se "encienden" solo cuando la entrada persiste durante un período mínimo de tiempo. Si el conjunto de soluciones para un problema particular es lo suficientemente pequeño, debería ser posible encontrar más analogías entre sistemas artificiales y células y establecer una tabla de motivos frecuentes con sus funciones. Incluso hay llamadas a la verificación de estas reglas mediante la construcción de redes mínimas de procesamiento biológico, con el uso de una “biología sintética” como prueba definitiva de comprensión[92]. Sin embargo, debe enfatizarse que las redes y sus motivos en los sistemas vivos tienen sus propias especificidades, porque a menudo evolucionaron para desempeñar múltiples roles y trabajar en entornos inestables[93]. Sin embargo, muchos artefactos modernos no están dominados por una función mínima, sino por la construcción modular, lo que garantiza la solidez y una mayor capacidad de evolución, por lo que se podrían generar más similitudes con los sistemas vivos en evolución descubierto en el futuro. Los lenguajes de la biología celular modular y la biología celular molecular son complementarios, porque los mismos motivos funcionales estudiados por la biología modular pueden ser implementados por muchos agentes moleculares diferentes: la biología celular está en transición desde una ciencia que se preocupaba por asignar funciones a proteínas individuales o genes, a uno que ahora está tratando de hacer frente a los complejos conjuntos de moléculas que interactúan para formar módulos funcionales[94].
3.16 Células en los tejidos: mecanismos moleculares y modulares de morfogénesis
La biología contemporánea se está dando cuenta una vez más de la importancia de una vieja sabiduría de que los animales y plantas multicelulares no están compuestos de células como un ladrillo, sino que los tejidos forman dominios especializados por crecimiento y división celular. y diferenciación. Además de centrarse en la actividad celular individual en este proceso, se debe considerar el todo integrado dinámico del organismo que produce y controla las células. Al igual que en la biología celular, se han hecho intentos para comprender los sistemas de desarrollo multicelulares en términos de las redes de procesamiento de información de las vías de señalización y la regulación de la expresión génica[95]. También se entiende que, junto con los módulos reguladores incorporados en las interacciones proteína-proteína y las estructuras promotoras de genes, es necesario tener en cuenta la forma dinámica del tejido. Por ejemplo, los gradientes de moléculas de señalización se remodelan dinámicamente por cambios en la forma del tejido[96]. Por lo tanto, cada tipo específico de célula dentro de un organismo puede entenderse completamente solo dentro del contexto de su posición específica dentro de un tejido y su función. Los enfoques modulares y moleculares ascendentes deben complementarse con conceptos descendentes que tengan en cuenta la estructura de los tejidos en desarrollo[97]. Comprender la naturaleza modular e interconectada de los sistemas vivos ha permitido revivir el concepto supracelular de campo en la biología del desarrollo y su reformulación en un marco compatible con la biología molecular. Tales campos modulares, que muestran tanto autonomía como jerarquía e interactúan entre sí, se han propuesto como mediadores entre el genotipo y el fenotipo tanto en la ontogenia como en la evolución. A diferencia de algunos conceptos de campo tempranos, estos campos se basan en interacciones genéticamente definidas entre células. Su jerarquía y establecimiento están influenciados por la información genética, pero el concepto de campo permite un cambio de enfoque al nivel supracelular de organización[98]. Durante mucho tiempo, el ECM[99] se consideró un material pasivo que llenaba el espacio entre las celdas[100]. Ahora se entiende como un depósito dinámico de moléculas de señalización. El ECM puede inhibir o facilitar la propagación de la señal[101], así como almacenar los morfógenos y liberarlos tras la degradación proteolítica o estimulación por señales adicionales. Las celdas móviles reorganizan la estructura y la posición de ECM rastrea la dirección de impulsión de la migración celular[102]. Las acciones de las células y la ECM son, por tanto, bidireccionales y complementarias. Más de un siglo después de que Roux definiera un programa de mecánica del desarrollo, los conceptos mecánicos se están convirtiendo en el sello distintivo de la biología del desarrollo convencional. Un programa ridiculizado por los genetistas del desarrollo temprano por no haber logrado ningún conocimiento mecánico ahora funciona completamente dentro del marco de la biología molecular. La biología del desarrollo también puede centrarse en los aspectos mecánicos del desarrollo como resultado de avances tecnológicos como pinzas ópticas[103], ablación con láser de células seleccionadas dentro del tejido[104] y microscopía de fuerza atómica para medir cuantitativamente la propiedades mecánicas de las superficies de la célula/ECM a una resolución de microescala[105]. Un número creciente de estudios ha demostrado cómo la señalización mecánica dentro de las redes de ECM celular interconectadas regula fuertemente el crecimiento, la expresión génica y la diferenciación[106], incluidos los aspectos mecánicos de la regulación de la invasividad celular en el desarrollo normal y en el establecimiento del cáncer.
3.17 Perspectivas sobre la estructura del citoplasma en el siglo XXI
Junto con la tradición establecida de asociar los procesos celulares con los orgánulos unidos a la membrana, los intentos de comprender la estructura y las propiedades del citoplasma han resurgido 100 años después del declive de los conceptos protoplásmicos, como bien se expresa en una cita de T. Mitchison: nada personifica más el misterio de la vida que la organización espacial y la dinámica del citoplasma[107]. La fase acuosa del citoplasma no es una bolsa de enzimas que se difunden libremente, como a menudo se percibe erróneamente a la luz de la bioquímica clásica, sino que está repleta de macromoléculas. El transporte difuso y la partición de macromoléculas y orgánulos en el citoplasma están muy restringidos por impedimentos estéricos y por interacciones de unión inesperadas[108]. Se cree que la alta viscosidad y el apiñamiento juegan un papel importante en la movilidad de los componentes citoplasmáticos. Las mediciones de la movilidad mediante técnicas modernas muestran un comportamiento diferente de la mera difusión pasiva. Curiosamente, las proteínas pequeñas a menudo se mueven más rápido que las moléculas inertes. Las interacciones débiles con los componentes citoplasmáticos circundantes posiblemente mejoren su movilidad. Los avances recientes han acumulado pruebas suficientes de la existencia de compartimentos sin membrana o "desnudos" en el citoplasma. Dichos compartimentos están formados por interacciones débiles multivalentes entre dominios repetidos de baja complejidad y/o dominios hidrófobos distorsionados[109]. La auto-interacción de los dominios asegura la separación de fases de los componentes del resto del citoplasma. Tras la formación de tal compartimento por proteínas polivalentes que interactúan, las parejas monovalentes que interactúan pueden entrar en el compartimento y concentrarse allí. Las gotitas sin membrana podrían desempeñar un papel en la concentración de componentes de una vía celular sin la necesidad de una barrera de membrana u otra jaula. Las gotitas individuales del mismo tipo pueden dividirse y fusionarse, y los componentes se intercambian constantemente con el grupo soluble[110]. Por lo tanto, estas estructuras poseen un alto nivel de dinámica interna y se caracterizan por un comportamiento similar al líquido, como el goteo, la fusión, la humectación y la capacidad de deformarse reversiblemente al encontrar una barrera física. Las gotitas de diferentes tipos (cada una basada en un dominio de auto-interacción diferente) pueden coexistir dentro del citoplasma sin mezclarse. Muchos de estos compartimentos son gránulos de ribonucleoproteína que consisten en moléculas de ARN multivalentes largas y proteínas de unión a ARN específicas. La formación de compartimentos sin membranas depende de la condición, es reversible y está controlada, incluida la modificación postraduccional. El entorno de estos compartimentos está aún más abarrotado que el resto del citoplasma. La combinación de separación de fases y apiñamiento molecular puede incluso atrapar proteínas con un número de copias extremadamente bajo[111]. Los efectos del hacinamiento sobre la dinámica de las vías de señalización, las redes de regulación genética y las redes metabólicas aún no se comprenden bien, pero el hacinamiento altera la difusión de proteínas y la cinética de las reacciones bioquímicas (debido a cambios entrópicos), a menudo en dependencia no lineal de las concentraciones de moléculas involucradas. Algunas de las ideas que involucran la separación de fases acuosas como un mecanismo de autoorganización se remontan a 1899 o posiblemente antes. E.B. Wilson propuso a fines del siglo XIX que los compartimentos no unidos a la membrana, como los gránulos P y los cuerpos de Cajal, podrían explicarse mediante los principios de la química coloidal[112]. También se conocen desde hace algún tiempo los cuerpos proteicos sin membrana de organización cristalina o cuasicristalina, probablemente formados por autoensamblaje. Las cáscaras de dichos compartimentos son permeables a los metabolitos pequeños, pero por lo demás mantienen el interior aislado del resto del citoplasma[113]. La mayoría de estas estructuras se descubrieron en células bacterianas, pero también se han descrito ejemplos de eucariotas. Además de la polimerización bien conocida de actina y tubulina en fibras citoesqueléticas, algunas enzimas metabólicas como la CTP sintasa también tienden a formar fibras. Las pantallas de microscopía de fluorescencia a gran escala revelaron la localización de muchas proteínas de levadura supuestamente citoplasmáticas en las fibras. Los estudios evitaron artefactos de sobreexpresión y fueron respaldados por métodos adicionales como la espectrometría de masas para candidatos seleccionados. El empaquetamiento de muchas proteínas en estructuras aún no caracterizadas se está volviendo evidente. Se han propuesto varias funciones para las fibras y los focos de proteínas, incluida la regulación alostérica eficaz, el blindaje de los intermediarios metabólicos y su canalización hacia vías complejas, y el almacenamiento de proteínas inactivas. Cada una de estas funciones ha sido demostrada en casos particulares pero, para la mayoría de las proteínas, se desconoce el impacto del ensamblaje en agregados y el impacto de la estructura altamente organizada de la estructura.
El citoplasma actualmente no está bien documentado o comprendido. Sin embargo, está claro que ciertas propiedades fisicoquímicas emergentes del interior de la célula no pueden ser reveladas por experimentos reduccionistas con algunos componentes aislados. Un desafío para la bioquímica posreduccionista es estudiar los fenómenos bioquímicos lejos del equilibrio químico y en condiciones fisiológicamente relevantes (es decir, dentro de las células, en extractos celulares complejos o en soluciones apiñadas[114]).
3.18 Investigación de lípidos y membranas en el siglo XXI
Aunque se está acumulando el apoyo a la existencia generalizada de compartimentos sin membrana en las células, la investigación moderna también demuestra el papel vital de las membranas biológicas. En interacción con los componentes citoplasmáticos, las membranas expanden los mecanismos de compartimentación celular y regulación funcional con capas adicionales de complejidad. Los lípidos, aunque anteriormente se pasaban por alto como meros componentes pasivos de las membranas, ahora se aprecian como determinantes cruciales de las propiedades de la membrana a diferentes escalas y son un tema de investigación clave en la biología celular moderna[115]. Los análisis lipidómicos mejorados demuestran que la diversidad de lípidos podría coincidir con la diversidad de especies de proteínas en una célula eucariota y que el catálogo de diversidad de lípidos aún se está expandiendo[116]. Un año después de la formulación del modelo de mosaico fluido de membranas plasmáticas, se planteó la hipótesis de que existen dominios más estables dentro de membranas uniformemente mezcladas[117]. Esta hipótesis de la "balsa de lípidos", basada en extracciones bioquímicas que indican compartimentos estables enriquecidos con esfingolípidos y esterol dentro de las membranas, nunca fue completamente aceptada. Sin embargo, el conjunto ampliado de herramientas computacionales, biofísicas y bioquímicas, que incluye simulaciones de dinámica molecular y métodos espectroscópicos avanzados, está conduciendo a una mejor comprensión de la heterogeneidad de las membranas. en diferentes niveles espaciales y temporales. Al igual que las macromoléculas en el citoplasma, los componentes de la membrana muestran una difusión anómala y se agrupan[118]. Los dominios autoorganizados transitorios impulsados ??por la segregación de componentes se informan a escalas desde unas pocas moléculas hasta micrómetros. Además, el citoesqueleto de actina cortical obviamente afina la organización de los microdominios, no solo actuando como un límite para la difusión de proteínas de membrana, sino también al influir en la organización de los lípidos y la transición de fase, que puede ser facilitada o suprimida aún más por la actina (dependiendo de otras condiciones específicas). Se ha observado la existencia de una fina red de actina-espectrina en los glóbulos rojos y recientemente se demostró en neuronas con la ayuda de microscopía de superresolución[119], lo que indica un fenómeno celular general. También son posibles reordenamientos locales rápidos de los dominios como resultado de la retroalimentación entre la composición local de fosfoinosítidos y el citoesqueleto de actina. Al igual que la corteza citoplasmática, se cree que la ECM influye en la movilidad de las proteínas de la membrana, lo que se ha demostrado en el caso de la limitación selectiva de la movilidad de las proteínas de la membrana plasmática vegetal por la pared celular. Las diferencias en la composición de lípidos locales regulan la función de las proteínas de la membrana, y una fracción sustancial de los lípidos de la membrana se une a las proteínas transmembrana en forma de una capa de solvatación hidrófoba en lugar de moverse libremente dentro de la bicapa[120]. Los efectos de la composición de lípidos sobre las propiedades físicas de una membrana son complejos y difíciles de predecir. Por ejemplo, el colesterol puede aumentar o disminuir la fluidez de la membrana local dependiendo de los otros componentes[121]. Las herramientas computacionales y experimentales ahora permiten evaluar el efecto de composiciones específicas sobre las propiedades físicas de la membrana y la estructura de las proteínas en diferentes situaciones. Una vez que las proteínas citoplasmáticas se reclutan en la membrana, la dimensionalidad de su movilidad se reduce de tres a dos dimensiones, aumentando su concentración efectiva en órdenes de magnitud. Por lo tanto, las membranas sirven como plataformas de interacción para las proteínas, que pueden ajustarse aún más al segregar a los socios de interacción en microdominios específicos. Ahora también se entiende que las membranas sirven como condensadores sintonizables para la integración y el almacenamiento de información en forma de acumulación de especies específicas de fosfolípidos de señalización[122]. La detección coincidente de más especies de lípidos, o un lípido específico junto con un compañero de interacción de proteínas, regula la unión de proteínas a los microdominios y membranas de diferentes orgánulos. Los mapas de interacción proteína-proteína a gran escala ahora se complementan con pantallas de alto rendimiento que prueban las interacciones proteína-membrana y su dependencia de la composición compleja de la membrana y propiedades biofísicas como la curvatura. Los efectos dinámicos de la composición de lípidos en los procesos celulares han sido difíciles de estudiar, porque la composición de la membrana está sujeta a una regulación estricta y rápida en forma de modificación del grupo de cabeza de fosfolípidos, transferencia de cadena de ácido graso y movimiento de lípidos entre las valvas de la membrana[123]. Además, las proteínas de transferencia de lípidos en relación con los sitios de contacto con la membrana se están estudiando como vías reguladas para el transporte de lípidos. Tal mecanismo de transporte es posiblemente mucho más rápido que el transporte vesicular, anteriormente considerado como el principal agente del movimiento de lípidos entre compartimentos[124]. Tecnologías emergentes como la activación optogenética de enzimas modificadoras de lípidos[125] y la fotoactivación de fosfolípidos enjaulados ahora permiten monitorear el efecto de los cambios de composición de la membrana en los procesos celulares a escala fisiológica espacio-temporal.
3.19 Hacia lo desconocido: el futuro de la biología celular
Además de la creciente resolución y cobertura de las mediciones moleculares, se ha logrado el descubrimiento de algunos componentes y mecanismos fundamentales previamente desconocidos. El descubrimiento de la interferencia del ARN en la década de 1990 reformó la percepción de la regulación de la expresión génica y fomentó un interés creciente en las especies de ARN no codificantes[126]. También hay factores que probablemente tengan un gran impacto pero son difíciles de medir y factores cuya existencia ni siquiera sospechamos, la verdadera materia oscura de la biología celular. Ejemplos de lo primero son las propiedades de proteínas intrínsecamente desordenadas, pequeñas especies de ADN intracelular e intercelular, interacciones débiles imposibles de detectar usando métodos bioquímicos tradicionales y distribución intracelular de especies de iones. Los últimos factores podrían ser motivos de interacción proteína-proteína no descubiertos, fases exóticas, tipos no detectados de moléculas pequeñas que existen en un número bajo de copias, modificaciones postraduccionales desconocidas o nuevos modos de comportamiento colectivo de biomoléculas. Con la mejora adicional de las herramientas disponibles, es posible que se revivan conceptos previamente abandonados y posiblemente olvidados en el marco de la biología molecular, como sucedió con las teorías endosimbióticas y la epigenética. Los biólogos celulares seguirán utilizando la combinación de enfoques de arriba hacia abajo y de abajo hacia arriba. La caracterización mecanicista detallada de los componentes individuales se combinará con enfoques a nivel de sistemas a gran escala, lo que permitirá la identificación de nuevos módulos celulares funcionales. El futuro de la biología celular (y de la biología en su conjunto) también radica en capturar los procesos de la vida simultáneamente a diferentes escalas espacio-temporales y la integración de los resultados en modelos multiescala, de modo que se pueda comprender la relación entre las interacciones de los componentes individuales y los fenómenos emergentes colectivos.
3.20 Modelo de división celular en eucariotes

Fig. 3.1. Modelo de división celular en eucariotes.
Las células pueden encontrarse en una de cuatro etapas del ciclo celular (ver Fig. 3.1) Fase G1 (Gap 1): intervalo entre la mitosis y la replicación del ADN, se caracteriza por el crecimiento celular. En G1 existe un momento crítico en el que la célula puede salir o permanecer detenida su proliferativo en G0. La prosecución de G1 depende de la presencia de señales favorables como mitógenos o factores de crecimiento. Entonces en presencia de estas señales, la célula entra en la fase S (síntesis) en donde ocurrirá la replicación del ADN. A este período le sigue la fase G2 (Gap 2) durante la cual aparecen eventos de transformación en la arquitectura celular, como la ruptura de la membrana nuclear, la condensación de la cromatina y la reorganización del citoesqueleto. El ciclo celular culmina con la segregación de los cromosomas duplicados en las células hijas durante la fase M (mitosis).
Entre los principales hallazgos que han dado forma al campo del conocimiento de la división celular, el exordio se da con el trabajo del biólogo celular alemán Theodor Boveri, Boveri fue el primero en acuñar los términos centrosoma y centriolo, y ya en 1887 se determinó que los elementos de la herencia y hereditarios estaban íntimamente relacionados con los cromosomas. Los centriolos son estructuras con forma de barril que son esenciales para la formación de los centrosomas, los cilios y flagelos. La desregulación de los números del centrosoma ha sido propuesta para contribuir a la inestabilidad del genoma y la formación de tumores, mientras que las mutaciones en las proteínas centrosomas, recientemente se han relacionado genéticamente con microcefalia y enanismo[127]. No es hasta 1951 que escribió Gunnar Ostergren en su trabajo seminal en Hereditas[128]; un estudio que arroja luz sobre uno de los procesos fundamentales de la biología celular, pero que había desconcertado a los científicos durante décadas, el movimiento de los cromosomas en la meiosis, Ostergren lo llamó polarización mitótica y cinetocoros no polarizados. Los cinetocoros son una estructura proteica donde se anclan los microtúbulos durante la mitosis y la meiosis.
En 1953 James Watson y Francis Crick hacen la descripción de la estructura del ADN y con ello abrieron la puerta a explicar la replicación del ADN; en 1950, la idea de que los cromosomas están hechos de ADN se convino en general, Hewson Swift la consideró un componente esencial de la genética. Sin embargo, el número de moléculas por cromosoma y la naturaleza de su régimen sigue siendo un misterio hasta ese momento. Esta duplicación del ADN se estudió con más detalle por Alma Howard que puso de manifiesto que la síntesis de ADN sólo se produce dentro de un determinado período de tiempo limitado - el medio de la interfase - al principio del proceso de división celular[129]. Este descubrimiento en última instancia condujo al modelo de división del ciclo celular eucariota en las fases S , G1, G2 y M.
Un gran avance se produjo en 1968, cuando Weisenberg y sus colegas identificaron la tubulina como la subunidad de la proteína de los microtúbulos[130]. Para ello, se aprovecharon de la afinidad de la colchicina - un inhibidor conocido de la mitosis - para una proteína asociada con el huso mitótico y los microtúbulos. Este descubrimiento finalmente proporciona la base necesaria para estudiar la organización de microtúbulos en el nivel molecular. Sin embargo, se desconocía exactamente cómo actuaba la tubulina polimerasa en los microtúbulos, en 1984 se introduce el concepto de inestabilidad dinámica, propuesto por Tim Mitchison y Marc Kirschner[131]. Estos investigadores proponen una serie de ensayos in vitro de microtúbulos de polimerización que les permitió medir la longitud y el número de microtúbulos individuales en lugar de las poblaciones a granel. Para su gran sorpresa, encontraron un estado que acuñaron como la "inestabilidad dinámica". Para explicar sus resultados, los autores propusieron que la mayoría de los microtúbulos crecen lentamente en el estado estacionario, mientras que una minoría se reduce rápidamente, permitiendo que la masa neta de polímero permanezca constante. Las transiciones entre el crecimiento y la contracción son poco frecuentes, pero pueden ocurrir en un amplio rango de concentraciones de tubulina, en consonancia con la observación de estos autores de que los microtúbulos individuales crecen de forma transitoria, incluso en concentraciones por debajo de la concentración de tubulina en estado estacionario. La inestabilidad dinámica gobierna el crecimiento de los microtúbulos de los centrosomas. La única diferencia es que en los centrosomas, se nuclean los microtúbulos de tubulina en concentraciones que están muy por debajo de la concentración en estado estacionario. Pero para empezar siquiera a comprender los centrosomas, los científicos tuvieron que esperar hasta 1989, cuando Oakley y sus colegas descubrieron la tubulina-Y sus propiedades de microtúbulos nucleantes[132]. Datos recientes han revelado que la familia de proteínas de tubulina es mucho más grande de lo que se pensaba. Seis familias distintas dentro de la tubulina se han descubierto y podría descubrirse más[133].
Para 1970 se identifica la existencia de que el ciclo celular podría depender de un conjunto de genes, los llamados genes de ciclo de división celular o cdc genes, cuya función es requerida en los puntos de tiempo específicos en el ciclo[134]. Para 2001 ya se habían identificado más de 100 genes cdc por Leland H. Hartwell y colegas, trabajo que le valió ser reconocidos por el premio Nobel de Medicina y Fisiología del año 2001. Uno de esos genes, el CDC28, o gen de inicio, controla la primera etapa en la fase G1. Las investigaciones de Hartwell también aclararon ciertos mecanismos de defensa del ciclo celular contra la irradiación o contra la alteración del orden correcto entre las diferentes fases del ciclo. Sin embargo, en 2010 se ratifica que la acción de control de los genes cdc no está sola, se ha identificado la interrupción de la multiplicación celular por campos eléctricos oscilantes[135].
En resumen, hemos visto hasta aquí, que la división celular se produce por una serie ordenada de cambios metabólicos y de morfogénicos que se denominan colectivamente el ciclo celular. Como ya hemos mencionado, este ciclo se divide en cuatro fases distintas. La replicación de los cromosomas se produce en la fase S (síntesis), mientras que la división celular se lleva a cabo en la M (fase mitótica). La brecha (G) entre la finalización de la fase M y el comienzo de la fase S se denomina la fase G1, y el período entre el final de la fase S y el comienzo de la fase M se llama la fase G2. Es importante destacar qué mecanismos de control actúan para regular el inicio de cada fase y para evitar que las transiciones entre las fases sean incorrectas. Las claves importantes sobre la naturaleza de la regulación del ciclo celular se establecieron por Rao y Johnson en 1970, por una serie de señales químicas que pueden difundirse libremente entre el núcleo y el citoplasma[136]. La identificación de estos factores a nivel bioquímico ha sido un foco principal de la investigación científica desde entonces.
Durante la mitosis y la meiosis en el ciclo celular se detiene, en los puntos de tiempo determinado, por ejemplo, cuando las células de los animales esperan la fertilización. Hasta 1971 Yoshio Masui y Clemente Markert identificaron el factor de promoción de la maduración (MPF). A continuación, tomó otros 17 años antes de que este factor fue purificado por Manfred Lohka, Marianne Hayes y James Maller[137]. Se determinó que MPF es también el factor clave que regula el inicio de la mitosis en todas las células eucariotas y que ahora se conoce como el factor de promoción de la mitosis. Es interesante pensar que, 40 años después del primer artículo mencionado MPF, la mayoría de los objetivos de este complejo son aún desconocidos[138].
En la década de 1970, la naturaleza y duración de la fase G0 eran una fuente de controversia, de hecho, algunos propusieron incluso que G0 en realidad no existía. En 1974, sin embargo, en la presentación de informes Actas de la Academia Nacional de Ciencias, Arthur Pardee argumentó que las células no entran en un estado G0 y que se trata de un estado equivalente con independencia de los medios por los que se induce. Él demostró que las células reentran en el ciclo celular en tránsito, una restricción -después de que se han comprometido con el ciclo celular- y argumentó que este interruptor está defectuoso en las células cancerosas y se activa cuando las condiciones no son óptimas para el crecimiento celular[139].
Las células siempre se dividen exactamente en el mismo tamaño. En 1975 se puso a la luz esta regulación estricta de la división mitótica, encontrando que depende de dos fuerzas opuestas que controlan la actividad de las quinasas (o cinasas) dependientes de ciclinas (Cdk), por cdc2[140]. Durante la mitosis y la meiosis, todos los cromosomas duplicados deberán separarse para las células hijas. Este proceso se basa en una porción del cromosoma mitótico donde las cromátidas hermanas se unen, conocido como el centrómero. Pero no fue hasta 1980 que pudo calificarse el papel de los centrómeros, cuando Louise Clarke y John estudiaron la levadura en ciernes[141].
En 1983, Evans, T. estudió la síntesis de proteínas en el erizo de mar. Descubrió que estaban presentes varias proteínas acumuladas a un alto nivel durante la interfase, para declinar abruptamente poco antes de que los huevos se dividían. Se acuñó el nombre de ciclinas para estas proteínas que mostraron comportamiento en el ciclo celular[142]. Posteriormente, las ciclinas se identificaron en una variedad de organismos, lo que demuestra la universalidad de este fenómeno[143].
Todas las células vivas deben replicar su ADN una vez y sólo una vez antes de dividirse, y deberán hacerlo con una precisión implacable para garantizar que su herencia genética se mantenga intacta. Los estudios clásicos a partir de 1987 en sistemas bacterianos y virales nos han formado en la idea de que la replicación del ADN consiste en complejos de la ADN polimerasa, que se mantienen bidireccionalmente desde un punto fijo - el origen de replicación[144]-. Las células eucariotas poseen normalmente varios cromosomas grandes, lineales, la réplica de la que es probable se plantea grandes problemas logísticos adicionales y topológicos, pero el ADN y los componentes de proteína implicada en la determinación de los pasos de la replicación del DNA eucariótico han sido definidos poco a poco en las últimas dos décadas[145].
Las células deben estar protegidas de una andanada de ataques con el fin de sobrevivir. Cada día se encuentran productos químicos y rayos ultravioleta que dañan su ADN y puede conducir a la muerte. ¿qué es lo que las protege de este mundo peligroso? La respuesta es sorprendentemente simple, las células poseen puestos de control de detención en la G2, por lo que les da tiempo para reparar los daños. El primer paso en la elucidación de esta vía fue tomada en 1988 por Ted Weinert y Hartwell Leland[146], con el descubrimiento de RAD9[147]. La proteína de punto de control del ciclo celular RAD9A es una proteína codificada en humanos por el gen RAD9A, requerida para que se produzca un alto total del ciclo celular[148].
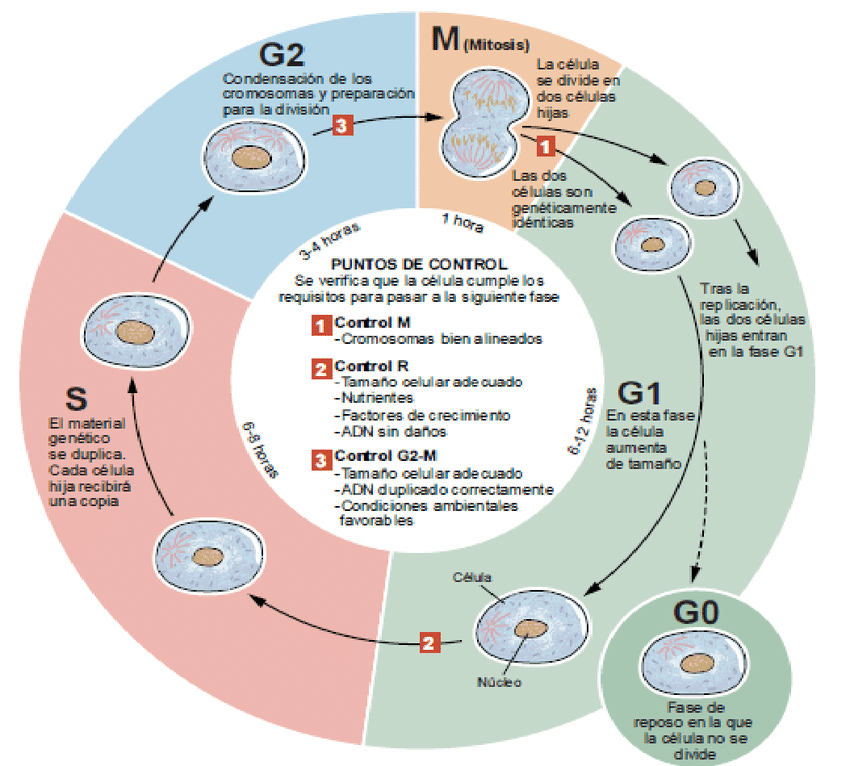
Figura 3.2. EL CICLO CELULAR. Tomado de noticias.cun enero-marzo 2007. TIMOTHY HUNT, NOBEL DE MEDICINA 2001
El gen del retinoblastoma, (RB), fue el primer gen supresor de tumores que se identificó en los niños con la hereditaria retinoblastoma. Su producto proteico, RB, se declaró más tarde ser funcionalmente inactivo en varios otros tipos de tumores humanos. Durante la década de 1980 y principios de 1990, RB fue identificado como un objetivo celular para oncoproteínas virales y los pasos significativos realizados en la comprensión de la función de RB en la regulación de la proliferación celular y la formación de tumores. Para los estudios del ciclo de división celular la familia de la vía de RB, así como las señales que inciden en la misión de fomentar o bloquear la proliferación celular se han vuelto de relevancia científica para la salud humana. Es decir, una de las funciones principales de Rb es la inhibición de la progresión del ciclo celular antes de la entrada en mitosis, de manera que la célula no entra en división hasta que está preparada para ello y se dan las condiciones adecuadas: Rb impide por tanto la proliferación celular[149].
A lo largo de la década de 1980, las supresiones y las mutaciones en TP53 ( el gen que codifica a p53) se informó de que un p53 defectuoso podría permitir que las células anormales proliferen dando por resultado cáncer. Baker y colegas llegaron a la conclusión de que las células en las fases pre-malignas de la progresión del tumor, tales como las células del adenoma, son menos sensibles a los efectos inhibidores del p53 en las células malignas. Esto condujo a la hipótesis de que las alteraciones genéticas que ocurren durante la progresión de los tumores colorrectales podrían aumentar la sensibilidad de las células para la inhibición de p53, p53 es un factor limitante de la velocidad de progresión del tumor[150]. El gen p53 también llamado el "guardián del genoma", se encuentra en el brazo corto del cromosoma 17 y codifica un factor de transcripción nuclear de 43.7 KDa[151].
Las células no son islas aisladas del cuerpo, cómo pocas células eucariotas se dividen de forma aislada, es imprescindible saber qué regula, cuándo y dónde las células se dividen en todo el organismo durante el desarrollo. ¿Cómo, por ejemplo, es la división celular, junto a eventos como la determinación de células destinas? Durante la década de 1980, el trabajo de Bruce Edgar y Pat O'Farrell hizo avanzar nuestro entendimiento en la mosca de la fruta Drosophila melanogaster, dado por stg ARNm[152]. Durante el ciclo celular, una célula se somete a una serie de eventos secuenciales -un paso tiene que ser completado antes de que el próximo se inicie. Esta progresión lineal se puede lograr de dos maneras: cada paso puede someter un producto como el paso anterior para su iniciación, o puede haber mecanismos de regulación de retroalimentación que garanticen que un paso posterior en el ciclo celular no se inicia sin un acontecimiento crucial. Conocido como puesto de control, en 1991, dos estudios publicados en la revista CELL, dirigidos por Andrew Hoyt y Andrew Murray, identificando el puesto de control del huso, mostraron la existencia de un mecanismo de retroalimentación de control que evita que las células abandonen la mitosis si su huso mitótico se ha reunido incompletamente[153].
La progresión a través del ciclo celular es altamente regulada. En todas las células eucariotas, la entrada en fase M está regulada por un complejo de la cinasa dependiente de ciclina (CDK) cdc2 y ciclina B (también conocida como MPF). La entrada en la fase S se regula de forma similar tanto en la fisión y la levadura en ciernes. Pero en 1991, un mecanismo diferente fue descubierto en los vertebrados, lo que lleva a la identificación de una nueva clase de proteínas[154]. Se estableció posteriormente que la ciclina E es -junto con la ciclina A- la subunidad reguladora de la quinasa CDK2 en la fase S. También se determinó que la ciclina D funciona como regulador de la CDK4 y CDK6 en G1, y en la regulación de la expresión de la ciclina E. Sin embargo , veinte años después de la publicación de este trabajo, la función exacta de ciclina C aún no se ha establecido[155]. Recientemente en 2010, se identifica que para CDK8 y sus socios ciclina C forman parte del complejo mediador que une a la maquinaria de transcripción basal para las proteínas reguladoras, están activas durante el desarrollo en respuesta a señales extracelulares y los niveles de estas proteínas son importantes para controlar la sincronización de los procesos de desarrollo[156].
En 1991 , cinco grupos de forma independiente informaron la identificación de una nueva clase de ciclinas, y otros dos grupos identificados relacionados con un CDC2 quinasa, CDK2. Juntas, CDK2 y estas nuevas ciclinas E, A, D regulan G1 y S como puesto de control en los vertebrados[157].
Conocemos desde 1971 que la actividad de promoción de maduración depende de un factor, más tarde identificado como ciclina B -CDC2 -, que oscila durante todo el ciclo celular, pero el controlador de estas oscilaciones tomó mucho más tiempo para ser identificado. El descubrimiento de que la proteólisis es un regulador clave de la ciclina, los niveles B mediados por un complejo ubiquitín ligasa grande, que ahora llamamos la anafase de promoción de complejo APC o ciclosoma; esto no sólo cambió nuestra visión del ciclo celular, sino que también tenía enormes implicaciones para entender la regulación de procesos celulares medidos por proteólisis ubiquitín. Aunque la mayoría las subunidades del APC han sido identificadas y sus funciones individuales reveladas. Pero a pesar de que la ciclina B fue una de las primeras proteínas que se reconoce con un objetivo regulado de ubiquitinación, (un proceso que nosotros ahora sabemos que es tan importante como la fosforilación en la regulación celular) la enzima que cataliza este proceso aún se queda con algunos de sus secretos[158].
En la década anterior a 1993, una ráfaga de actividad en el campo del ciclo celular ha dado lugar a la identificación de las proteínas quinasas dependientes de ciclinas (CDK) y sus asociados ciclina regulador. Estos complejos había demostrado su participación en la regulación de las transiciones del ciclo celular para permitir la replicación del ADN (fase S ) y la entrada a la mitosis.
El supresor de tumores retinoblastoma (RB). Esta proteína también entró en la ecuación, en la que había demostrado ser un importante objetivo para ciclina / CDK fosforilación, lo que lleva a la inhibición y, por tanto, a la progresión del ciclo celular. 1993 fue un año emocionante de la investigación convergente: varios grupos de forma independiente – y a través de vías muy diferentes - identificaron el primero de los importantes reguladores del ciclo celular , llamadas colectivamente los inhibidores de CDK: p21, p27Kip1,p53,p120[159].
Un hecho emocionante se dio en 1994, cuando Kim Nasmyth y sus colegas, trabajando en la levadura en formación Saccharomyces cerevisiae, mostró que la proteólisis también se requiere para que las células comiencen la replicación del ADN. La ubiquitín ligasa responsable de esto fue identificada posteriormente como SCF. Skp1 , Cdc53 y Cdc4, se reconoce y degrada Sic1. A diferencia de la APC, SCF reconoce específicamente las proteínas que son fosforiladas. Así que el SCF proporciona un vínculo entre los reguladores de proteínas que están implicadas en la fosforilación y la proteólisis reuniendo una vía central en la regulación del ciclo celular eucariota[160].
El momento de separación lo es todo en la división celular. Por ejemplo, después de que una célula en división se ha duplicado sus cromosomas, se necesita mucho cuidado de no separar las copias hasta que esté seguro de que la duplicación se ha realizado correctamente y que las copias han sido alineados correctamente. De lo contrario, cada célula hija recién formada se cargará con un complemento incorrecto del material genético. Los cromosomas se duplican en la fase S, pero los dos ejemplares (cromátidas hermanas) se mantienen juntos hasta mucho más tarde, la transición metafase -anafase - durante la mitosis. Sin embargo, como Frank Uhlmann del Imperial Cancer Research Fund de Londres dice: "Desde el punto de vista del cromosoma, parece una tarea paradójica, donde la primera aparece estrechamente vinculada a la copia que hermana, pero luego la deja ir, y en la célula hija pasó lo contrario". [lxix] ¿Cómo funciona esto? mostraron que Esp1 está obligado a destruir Scc1 en el inicio de la anafase. Descubrieron que Scc1 se divide en el inicio de la anafase pero no cuando está mutado Esp1. También genera una ancla Scc1, y encontraron que las células que expresan esta proteína no podía separar las cromátidas hermanas[161].
En 2010 es revelada la dinámica de adhesión célula a célula, gobernada por al E-cadherin y el p120, permiten modelar la división celular en el espacio tiempo corporal, revelando las uniones de estabilidad y dinámica celular.[162]
En resumen nuestro estudio comenzó en 1902 y concluye en 2010, intenta explicar la división celular desde a dentro, a continuación expresaremos los detalles más íntimos de este fascinante proceso esencial para la vida.
3.20.1 Interruptores del ciclo celular
El crecimiento de la célula humana es controlado por un juego de proteínas que actúan como interruptores moleculares. Estos interruptores se encienden o se apagan por la orden de varias proteínas y aseguran que la célula sólo crece cuando es apropiado. En el cáncer, las alteraciones genéticas causan a menudo la pérdida de restricciones de estos interruptores y llevan al crecimiento desenfrenado de la célula. Estos interruptores se les conoce como CDK’s.
En el estudio de eucariotes, CDK es parte de la maquinaria de la división celular, gran parte de los eventos del ciclo celular están regulados por las ciclinas, CDK (Cyclin Dependent Kinase) por ejemplo, los inhibidores de CDK4. Las ciclinas son proteínas cuya concentración y actividad varía en cada etapa del ciclo celular. Las CDK y sus inhibidores, que pueden ser de la familia de los KIP (Kinase Inhibitor Protein) o INK4 (inhibidor CDK4[163]), se encuentran en un nivel constante a lo largo del ciclo celular. Las ciclinas se unen a la quinasa, produciendo su activación; una vez activadas, éstas cumplen funciones específicas actuando sobre otras proteínas nucleares, lo que condiciona la progresión de la célula a otros estados del ciclo celular. Diferentes ciclinas están envueltas en distintas fases del ciclo y se sabe que todas ellas pueden ser blancos de cambios genéticos o pueden ser alteradas en el proceso de ontogénesis[164].
La pérdida de la sensibilidad a la inhibición del proceso de crecimiento es el resultado de la pérdida del control positivo de las ciclinas o pérdida de la función de los inhibidores CDK’s. Así también la inestabilidad génica, determinada por la habilidad de la célula tumoral de saltarse los mecanismos de restricción presentes al inicio de la fase S (fase de síntesis) del ciclo celular, conduce a la perpetración de una línea celular con DNA defectuoso. La sobreexpresión de ciclinas ha sido demostrado que genera cánceres como el de colon y mama, donde está desregulada la expresión de la ciclina E[165] y POM121 está involucrado en la fusión de las membranas nucleares interna y externa durante la interfase[166].
La entrada al ciclo celular, en las células de mamíferos es determinada por la actividad de las ciclinas CDK’s, que consisten en un centro quinasa y subunidades ciclinas asociadas. Para entrar en la fase S -la síntesis de ADN- los jugadores importantes son en G1 CDK4 y CDK6 que son los complementos con las ciclinas tipo D (D1-3), y CDK2 que es el complemento con ciclinas tipo E (E1 y E2[167]).
Se sostiene ampliamente que la ancha gama de células de mamíferos es producto de una serie de decisión –comandos- en la fase G1 de la división celular para proceder a través del ciclo celular; para la diferenciación y para cesar el crecimiento o división. Se prevé actualmente que las células toman una decisión en la fase de G1 del ciclo de la división al diferenciarse. Después de la diferenciación las células permanecen en la fase G1 y no se dirigen a iniciar la síntesis de DNA. Células que están sufriendo la diferenciación y que no entran en la fase S, G2, o M escalonada. Células que estaban detenidas en S, G2, y M escalonada cuando el comando de iniciación fue recibido atraviesa estas fases.
G0. La existencia de un punto de decisión en el proceso de una fase G1, provee soporte para la idea general de que allí reside un importante control del ciclo celular. La propuesta de un punto de restricción introduce a la célula en G0. Idea general de una fase de control G1 detenida, que soporta la idea de más de un comando para iniciar la diferenciación[168].
Por otro lado, puede suceder que las células cesen de proliferar, pero retengan la capacidad de dividirse posteriormente, es la denominada quiescencia celular, también se denomina a esta detención del proceso en G1 como fase G0 . Estas células pueden entrar de nuevo en fase G1 por estímulos externos y proliferar, por ello se denomina control externo del ciclo celular, se realiza por moléculas a las que se han denominado factores de crecimiento[169].
Los procesos de proliferación, diferenciación y apoptosis celular utilizan sistemas de transmisión de señales similares que en ocasiones incluso comparten sus componentes estructurales básicos. La señal, codificada en forma de factor difusible, interacciona y activa un receptor específico en la célula efectora. Como consecuencia, se pone en marcha un complejo sistema de transmisión de señales intracelulares que a través de diversas enzimas citoplásmicas, concluye en la activación de factores de transcripción específicos. Los principios básicos de este proceso son compartidos en la regulación de la proliferación, diferenciación y apoptosis.
El que una célula inicie la fase G1 está determinado por la proteína retinoblastoma (Rb) que une y atrapa al factor transcripcional E2F. Éste controla la síntesis de proteínas necesarias en G1. Esta inhibición desaparece cuando la CDK4 se activa y fosforila a Rb activándose la capacidad transactivante de E2F. La presencia de mitógenos reduce los niveles de la proteína p16 inhibidora de la CDK4 y a su vez induce la aparición de ciclina D, acciones que aumentan la actividad CDK4. Uno de los sustratos más importantes de esta kinasa es la proteina retinoblastoma (Rb). La CDK4 hiperfosforila a Rb liberando el factor transcripcional E2F. Este se une a secuencias específicas del ADN para promover la síntesis de enzimas requeridas en la síntesis del ADN (Polimerasas, Dehidrofolato reductasa y ciclinas E y A). La aparición de la ciclina E permite la activación de CDK2 que promueve la desaparición de su inhibidor p27 a través de fosforilación y activa la maquinaria de replicación[170].
En resumen, los actores principales del ciclo celular son las CDK’s, un grupo de serina / treonina quinasas que forman complejos heterodiméricos activos, son subunidades reguladoras. Varios CDK principalmente CDK4, CDK6, CDK2 y CDK3 cooperan para conducir las células a través de G1 a la fase S. CDK4 y CDK6 se implican en G1 temprana, mientras que CDK2 es necesaria para completar G1 e iniciar la fase S[171]. CDK4 y CDK6 forma complejos activos con las ciclinas de tipo D (ciclinas D1, D2 y D3[172]). Estas quinasas son muy relacionadas y, hasta ahora, se han resistido a la diferenciación funcional, a excepción de su patrón distinto de activación[173].
CDK reguladores. Al controlar el nivel de actividad CDK, reguladores CDK también puede controlar el bucle del ciclo celular. Estos incluyen activadores, principalmente las ciclinas, e inhibidores, conocidos genéricamente como CKIs (CDK inhibidores de quinasas). CDK también son reguladas por la fosforilación: deben ser fosforiladas en un residuo de treonina (T172 en CDK4 y T160 en CDK2) -situados en su bucle T- para la actividad catalítica adecuada[174]. Esto se lleva a cabo por el complejo ciclina-CDK7-H (también conocida como CAK; CDK-que activa la cinasa o también llamada quinasa), una serina / treonina quinasa que también está implicada en la transcripción y la reparación del ADN, fosforilación inhibitoria de treonina adyacentes y los residuos de tirosina (T14/Y15 en CDK1) está mediada por quinasas de doble especificidad (como WEE1 y MYT1[175]). Esta inhibición se alivia cuando la fosfatasa CDC25 (CDC25A, CDC25B y CDC25C) desfosforiliza estos residuos, lo que desencadena la entrada en mitosis.
CKI’s. CKI’s son de dos tipos. Los cuatro miembros de la familia de INK4 - INK4a (también conocido como P16), INK4B (también conocido como P15), INK4C (también conocido como P18) y INK4D (también conocido como P19) - ejercen su actividad inhibitoria al unirse a la CDK4 y CDK6 quinasas y la prevención de su asociación con las ciclinas de tipo D. Aunque las proteínas INK4 son bioquímicamente indistinguibles entre sí in vitro, la generación de ratones dirigida de genes que son deficientes para cada una de estas proteínas ha revelado importantes diferencias funcionales[176]. Los tres miembros de la WAF familia de KIP - WAF1 (también conocido como p21), Kip1 (también conocido como P27) y KIP2 (también conocido como P57) - forman complejos heterotriméricas con el G1 / S CDK[177]. Sin embargo, en cantidades estequiométricas, sólo inhiben la actividad quinasa de los complejos CDK2-ciclina E. CKI’s se inducen en respuesta a diferentes procesos celulares. Por ejemplo, WAF1 es uno de los efectores de P53, un supresor de tumores que es importante en el puesto de control de daños del ADN - un proceso crucial para el cáncer humano[178].
Los substratos de CDK. Los substratos primarios CDK4/6 y CDK2 en el desarrollo de G1 son los miembros de la familia de la proteína retinoblastoma - RB, P107[179] y P130[180]. [xci] , [xcii] regulan la expresión de las P130, P107 y E2F-4 en células humanas[181]. Estas moléculas “cercenan” los sitios para una serie de proteínas que deben regularse herméticamente a lo largo del ciclo celular. Por ejemplo, las proteínas de RB envuelven a la familia E2F de la transcripción, para asegurar que ellos permanezcan inactivos durante fase M y G0. Además, los complejos de RB-E2F participan en la represión activa de algunos promotores: un mecanismo que involucra a otras familias de proteínas, como las histonas deacetilasa y los remodelares cromosomáticos, complejos SWI/SNF[182]. Hasta ahora, más de 100 son las proteínas de RB que han sido identificadas[183], pero la relevancia fisiológica de la mayoría de estas interacciones no se ha establecido. RB también puede regularse por acetilación, por medio de las histonas acetilasa asociadas a P300/CBP[184]. Estos acetilasas están bajo el comando del ciclo celular y previenen la fosforilación de RB por CDK2-cyclin E[185].
Ciclinas tipo-D. Las ciclinas del tipo-D son integradores importantes de la señalización mitogénica pues su síntesis es uno de los puntos finales y principales de las vías RAS/RAF/MAPK[186]. Si la disponibilidad de suficientes cantidades de ciclinas tipo-D hace que las células se vuelvan independientes de estímulos mitogénicos. Para las células que carecen de ciclinas D1, éstas pueden proliferar indicando que esta molécula no es terminantemente necesaria para G1[187]. Las tres ciclinas tipo D, sin embargo, es evidente que la sobreexpresión ciclina D1 facilita el paso en G1 y promueve la proliferación de la célula. Ciclina D1. Además la ciclina D1 es esencial para formar tumores superficiales oncogénicos[188].
3.20.2 Mutaciones en el ciclo celular
El análisis molecular de tumores humanos ha mostrado que reguladores del ciclo celular frecuentemente deforman en neoplasia humana (ver figura 4), este análisis subraya como importante el mantenimiento del ciclo celular en la prevención del cáncer humano. Las alteraciones incluyen la sobreexpresión de ciclinas (principalmente D1 y E1) y CDK’s (principalmente CDK4 y CDK6), así como la pérdida de CKI (principalmente INK4A, INK4B y KIP1) y expresión de RB. Los cambios asociados a tumores en la expresión de estos reguladores frecuentemente son el resultado de las alteraciones del cromosoma (la amplificación de ciclina D1 o CDK4, permutación de CDK6 y supresión de proteínas INK4 o RB) o inactivación epignética (la metilación de INK4 o promotores RB) las mutaciones en CDK4 y CDK6[189].
 18.38.42.png)
Fig. 3.4.- La mutación de reguladores en G1 o S en el cáncer humano. Se han considerado sólo alteraciones que ocurren en más de 10% de los tumores. Los números representan el porcentaje de tumores con las alteraciones en cualquiera de los reguladores del ciclo-celular listados. Los sitios de especificidad genética o la alteración que causan tumores se ha definido en color rosa. Las alteraciones sin ninguna explicación mecánica se ha mantenido en amarillo. Las alteraciones pertinentes para la prognosis del tumor se indica por los asteriscos[190].
 18.40.51.png)
Fig. 3.5 Punto de control de restricción y tránsito G1-S.
Los avances recientes en biología molecular permiten agrupar a los genes del cáncer en varias categorías de acuerdo a sus funciones conocidas. Una visión simplificada reduce a dos, los tipos fundamentales de genes relacionados con la aparición del proceso tumoral: oncogenes y genes supresores[191].
Oncogenes. Su función normal es la regulación de las rutas de señalización de la proliferación celular, denominándose proto-oncogenes. Tras su alteración se activan de forma constitutiva, manteniendo la señal mitogénica permanentemente activa. Las funciones de cada grupo se pueden resumir siguiendo el esquema de funcionamiento de los genes normales de los que derivan, involucrados en la regulación de la transmisión de señales[192]. El diseño de nuevos antitumorales se basa precisamente en el bloqueo selectivo de su función mediante moléculas que interrumpan esta cascada de señales en cada uno de sus puntos conocidos. Los niveles de señalización y mensajeros primarios, fundamentalmente factores de crecimiento y hormonas.
Genes supresores. Son genes cuya función primordial es frenar la proliferación. Su función se realiza dentro del contexto de la regulación del ciclo celular, siendo la pérdida de su función durante el proceso de carcinogénesis, la que contribuye a la aparición de tumores al perderse el control sobre el freno de la proliferación. Se conocen una decena de genes supresores como retinoblastoma (Rb), P53, los genes de la neurofibromatosis tipo 1 y 2 (NF1, NF2), el gen de la poliposis adenomatosa colónica (APC[193]), el gen del síndrome de von Hippel-Lindau (VHL) y del tumor de Wilms (WT-1). Los genes mejor caracterizados son el gen del Rb y P53[194].
En el caso de Rb, su regulación se produce mediante un proceso de fosforilación/desfosforilación actuando como modulador de la función del factor de transcripción E2F-DP. La fosforilación de Rb está claramente asociada con su inactivación como proteína supresora de la proliferación. Cuando está hipofosforilada, en las fases G0 y G1 temprana, la proteína Rb secuestra al factor E2F impidiendo su actividad. Tras la fosforilación, que se produce en la fase media de G1 y se mantiene a lo largo de las fases S, G2 y gran parte de la M, Rb libera al factor E2F[195], permitiéndose su actividad transcripcional. La desfosforilación se produce en la salida de la fase M y entrada en la fase G0, secuestrándose de nuevo al factor E2F[196].
En conclusión parcial, observamos que la falla en los interruptores del ciclo celular, con frecuencia deriva en cáncer, es considerado como una enfermedad del ciclo celular. Aunque muchos mecanismos específicos de regulación del ciclo celular han sido estudiados en profundidad in vitro, aún no está claro como los diversos procesos celulares están desregulados con la división celular en el cáncer humano. Una visión integrada de los estudios bioquímicos y el análisis de las mutaciones del cáncer, sin duda, ayudará a explotar este conocimiento en los enfoques terapéuticos. La investigación futura exigirá explicar aspectos cruciales de la regulación del ciclo celular in vivo, como las vías de síntesis y proteólisis de proteínas esenciales, de control posteriores a la traducción y, sobre todo, la especificidad funcional o redundancia entre células familiares. Del mismo modo, explicar cómo las células coordinan el crecimiento celular y la proliferación celular, y cómo decidir si o no activar ciclo a ciclo la reparación celular, es un desafío aún mayor. En última instancia, es observable en la literatura referida que el diseño de fármacos contra el cáncer específico debe provenir de enfoques multidisciplinares que combinen, entre otras estrategias: patología molecular[197], para identificar todas las mutaciones/alteraciones presentes en los tumores humanos, genómica funcional[198], para seleccionar los objetivos más importantes, estructurales y biología computacional[199], para predecir la estructura de las proteínas y las interacciones de inhibidores; la química combinatoria asistida por ordenador[200], para el diseño de inhibidores y modelos animales con un valor muy predecible, para aumentar las posibilidades de éxito de los candidatos de la droga principal en la clínica.
3.20.2 Puntos de control
La E2F en G1/S, familia de factores de transcripción, determina si una célula se divide por el control de la expresión de los reguladores clave del ciclo celular. Las E2F’s individuales se pueden dividir en subgrupos distintos que actúan en oposición directa entre sí para promover tanto la proliferación celular o en ciclo celular, la diferenciación terminal de salida[201]. ¿Cuál es la base molecular de estos interruptores de control? y ¿cuáles son sus consecuencias biológicas?
El ciclo celular en mamíferos es un proceso altamente regulado que está influido por señales tanto positivas como negativas de regulación del crecimiento durante la etapa G1. Estas señales actúan en el control de la actividad transcripcional de un factor celular llamado E2F. La activación de E2F es suficiente para que la participación sea irreversible en las células al someterse a la replicación del ADN, por lo que E2F es crucial en el control de la proliferación celular en células normales y tumorales[202].
En la última década, la actividad de E2F se ha demostrado que surgen de la acción concertada de una familia de proteínas que tienen distintas propiedades biológicas de los demás. La forma E2F fue identificada y relacionada con el control del ciclo celular y el cómo la actividad del E2F individual está influenciada por su interacción con los miembros de la familia de proteínas de retinoblastoma.[203] Da lugar a papeles opuestos, tanto en la activación o inhibición de la proliferación celular.
E2F fue descubierta originalmente como una actividad celular que se requiere para la región 1A (E1A), la transformación de las proteínas de los adenovirus para mediar la activación transcripcional del promotor viral E2; E2F controla la transcripción de genes celulares que son esenciales para la división celular[204]. Estos reguladores del ciclo celular codifican por ejemplo, a la ciclina E, ciclina A, Cdc2, Cdc25A, la proteína retinoblastoma (pRB) y E2F1), enzimas que participan en la biosíntesis de nucleótidos y en los principales componentes de la maquinaria de replicación del ADN (Cdc6, ORC1 y en proteínas de mantenimiento minicromosoma (MCM)).
En una elegante serie de experimentos en la década de 1990, Joe Nevins y colegas deducen cómo E2F está regulado en las células normales, determinando el mecanismo de activación por E2F-E1A.[205] Factores de transcripción E2F desempeñan un papel crítico en el control de la progresión del ciclo celular, regulando la expresión de genes implicados en la replicación del ADN, la reparación del ADN, mitosis y el destino celular[206].
La asociación del factor E2F de transcripción con la pRb y proteínas p107 parecen regular la actividad de E2F y, a su vez , afectan a la progresión del ciclo celular. Esto los llevó a mostrar que E2F es inhibida por su asociación con pRB, una proteína conocida E1A-asociada y que estimula E1A a la entrada del ciclo celular por el pRB y la inducción de la liberación de E2F transcripcionalmente activa[207]. Al mismo tiempo, La Thangue identificó una actividad celular llamada factor de diferenciación de transcripción regulada 1 (DRTF1), durante la diferenciación de carcinoma embrionario F9 en células madre[208]. El sitio de unión al ADN de DRTF1 resultó ser el mismo que el de E2F, y DRTF1 también interactuó con pRB. Ahora está claro que DRTF1 y E2F son el mismo factor. Es importante destacar que la afinidad de DTRF1 en microsecuenciación condujo a la identificación de un componente clave de la llamada proteína DRTF1/E2F llamada DRTF proteína 1 (DP1[209]).
pRb fue el primer supresor tumoral en ser identificado, y está ausente o mutado en al menos un tercio de todos los tumores humanos[210]. En las células no transformadas, la capacidad de pRb para unirse a E2F está regulada por la fosforilación de su ciclo celular dependiente. pRB es hiperfosforilado durante G0 y G1 temprana, y se une e inhibe a E2F.
Factores de crecimiento mitogénico inducen a la activación secuencial de los complejos quinasa dependiente de del ciclo celular Cdk4/Cdk6-ciclina D y Cdk2-ciclina E, que luego de fosforilar pRB provoca la liberación de E2F. La activación resultante de los genes E2F durante el final de G1 parece ser suficiente para que las células inicien la replicación del ADN.
Ocho genes humanos han sido identificados como componentes de la actividad transcripcional E2F[211]. Sobre la base de la homología de secuencia y propiedades funcionales, estos genes se han dividido en dos grupos distintos: los E2F’s (E2F1 - E2F6), y las DP’s (DP1 y DP2). Los productos proteicos de estos dos grupos heterodimerizan para dar lugar a la actividad funcional E2F, y todas las combinaciones posibles de los complejos de E2F-DP existen in vivo[212]. La completa secuencia del genoma humano no ha detectado a genes E2F adicionales DP. Así, el reto ahora es comprender el papel específico de los complejos individuales E2F-DP.
La activación de E2F. El miembro fundador de esta subclase, E2F1, se clonó en virtud de su capacidad para unirse PRB[213]. E2F1 se demostró que se unen al ADN, de forma DP-dependiente, y el complejo resultante es un potente activador transcripcional de E2F promotor[214]. Posteriormente, dos grupos aislados ADN complementario de codificación de E2F2 y E2F3 humano[215]. Estos dos E2F son altamente homólogos a E2F1 - especialmente en los ámbitos que son responsables de unión al ADN, dimerización DP y PRB vinculante - y manifestaciones similares de unión al ADN y las propiedades de transactivación. Estudios han demostrado que el lugar E2F3 expresa dos distintos transcripciones[216].
La transcripción codifica las especies originales de E2F3 original, ahora designado E2F3A. La transcripción segundo, llamado E2F3B, se transcribe a partir de un promotor reconocido previamente en el primer intrón del E2F3A, y su producto es idéntico a la proteína E2F3A excepto que carece del dominio amino-terminal[217].
Las propiedades de E2F3B aún no se han descrito completamente. E2F1, E2F2 y E2F3a podría contribuir a la represión de los genes E2F-respuesta mediante PRB. Sin embargo, la determinación de la sobreexpresión y modelos de ratón mutante indican que el papel fundamental de estos tres E2F’s es la activación de genes que son esenciales para la proliferación celular y la inducción de apoptosis. En consecuencia, nos referimos a este subgrupo como el E2F activos.
Rol en el desarrollo normal. Cepas mutantes de ratón se han utilizado para investigar el papel de la activación en E2Fs en el desarrollo normal[218]. E2F1 y E2f3 en ratones mutantes mostraron problemas de desarrollo completamente diferentes[219]. Con una proporción significativa del E2f3-/-, los ratones mueren en el útero y la mayoría de los sobrevivientes adultos mueren prematuramente de insuficiencia cardíaca congestiva. Por ejemplo, la neurogénesis es un proceso altamente regulado por el cual los precursores neurales se dividen y diferencian, dando lugar a las células que producen el sistema nervioso. El retinoblastoma (Rb) supresor de tumores es un regulador clave de ciclo celular, junto con otros han demostrado jugar una serie de papeles en el desarrollo neurológico, incluyendo la proliferación, supervivencia y más recientemente, la migración neuronal. La familia E2F de factores de transcripción se compone de E2Fs 1 a 8 , sin embargo, E2F1, E2F2 y E2F3, la E2Fs llamadas activas, son la clave, objetivos de la interacción Rb- mejor conocido por su papel en la promoción de la progresión del ciclo celular. Ambos E2F1 y E2F3 son candidatos probables implicados en Rb- que median la regulación de la neurogénesis. E2F1 y E2F3 son funcionalmente relevantes en la proliferación de precursores neurales, células de salida del ciclo, laminar y patrones. Cada parte también interviene en la Rb requisito para la supervivencia neuronal. La migración neuronal, sin embargo, está específicamente mediada a través de E2F3, más allá de su papel en la regulación del ciclo celular.[220]
Por el contrario, E2F1-/- ratones son viables y fértiles, pero tienen diferentes tejidos específicos anormales[221], estos incluyen un exceso de células T, que resulta de un defecto en la selección negativa, y el desarrollo de la atrofia testicular entre los 9 y 12 meses de edad. Por último, y lo más sorprendente, los ratones con deficiencia de E2F1 desarrollar un amplio espectro de tumores que se presentan entre 8 y 18 meses de edad. Tres posibles mecanismos han sido propuestos para las propiedades de supresión de tumores de E2F1. Inicialmente, esto se pensó que era debido a la capacidad de E2F1 para participar en la represión de los genes E2F mediante la negociación de pRB[222]. Sin embargo, la ausencia de tumores en ratones que son deficientes para E2F4, el E2F principal in vivo, planteó sus dudas acerca de este modelo[223]. En la actualidad, muchos investigadores creen que E2F1 actúa como un supresor del tumor a través de su capacidad de inducir apoptosis[224]. Sin embargo, otros estudios indican que E2F1 también podrían estar implicados en la vía ADN respuesta a daños[225].
Lo más desconcertante es E2F1, se ha informado que asociado con NBS1 y la recombinación Mre11/complejo de reparación. Los autores proponen que E2F1 es necesario para el objetivo del complejo Mre11 cerca de los orígenes de replicación del ADN para suprimir el despido de estos orígenes cuando el ADN está dañado[226].
Una cuestión clave es preguntarnos si el E2Fs tiene diferentes funciones específicas o que se superponen. El campo ha comenzado a abordar esta cuestión mediante la generación de cepas de ratones mutantes que carecen de las combinaciones de los genes E2F1 y E2F2, cooperan en la regulación de la proliferación celular y la diferenciación hematopoyética y también en la supresión de tumores[227]. Así como sus funciones se superponen en la inducción de la proliferación y la apoptosis, E2F1, E2F2 y E2F3 parecen tener funciones fundamentales y se superponen en el desarrollo[228]. Por lo tanto, la supresión del tumor parece ser una propiedad específica de E2F1 y E2F2, pero no E2F3.
Las E2F’s represivas. La subclase segunda de la familia E2F es E2F4 y E2F5. Estos fueron identificados originalmente por el clonado en virtud de su asociación con p107 y p130[229]. En consonancia con esta observación, E2F4 y E2F5 están regulados de manera diferente desde el E2Fs activo in vivo. En primer lugar, niveles significativos de E2F4 y E2F5, se detectan en reposo (G0) a las células, mientras que E2F1, E2F2 y E2F3a se restringe fundamentalmente en células en división cativa[230]. En segundo lugar, se unen a las proteínas E2F subgrupos diferentes de bolsillo in vivo[231]. Considerando que la activación de E2Fs están específicamente reguladas por el pRB, E2F5 está regulado principalmente por p130 y E2F4 asociados con cada una de las proteínas en diferentes puntos del ciclo celular. Como E2F4 se expresa en niveles más altos que los demás miembros de la familia E2F, representa al menos la mitad de la pRb, p107 y p130-E2F actividad asociada in vivo.
Regulación por localización subcelular. En contraste con la activación de E2Fs, E2F4 y E2F5 estos son pobres activadores transcripcionales en ensayos de sobreexpresión, y no pueden conducir a las células en reposo a volver a entrar en ciclo celular[232]. La actividad diferencial de los dos subgrupos E2F resulta de las diferencias en su localización subcelular: E2F1, E2F2 y E2F3 son constitutivamente nucleares, mientras que E2F4 y E2F5 son predominantemente citoplasmática[233]. Estudios de mutagénesis han identificado la base subyacente de esta diferencia. El E2Fs activa una señal de base canónica de localización nuclear (NLS) en su dominio amino-terminal que es suficiente para mediar su localización nuclear[234].
Por el contrario, E2F4 posee señales hidrofóbicas de exportación nuclear (NES) y su localización citoplasmática depende del factor de exportación nuclear CRM1[235]. El DP-endógeno E2F4 y E2F5 DP-complejos son consistentemente detectado en el citoplasma, que indica que E2F4 y E2F5 no puede activar genes E2F in vivo, debido a su localización citoplasmática. Significativamente, la asociación con el pRB o p130 es suficiente para inducir la localización nuclear de estos E2Fs en vivo[236]. De hecho, en las células G0/G1, E2F4 y E2F5 se encuentra para la mayoría de los complejos nucleares E2F-DP-proteína. En estos complejos asociados con HDACs in vivo[237], E2F4 y E2F5 se cree que son cruciales para la mediación de la represión transcripcional de los genes E2F. La mutación de los sitios E2F vinculantes, con conocidos promotores E2F-sensibles (tales como B-myb, cdc2 y E2F1) conduce a la transcripción de estos promotores en G0/G1[238]. En ensayos in vivo se demuestra que la viruta en niveles significativos de E2F4, p107 y p130 ocupan los promotores de muchos genes E2F durante las etapas del ciclo celular[239]. A medida que avanzan las células a través del ciclo, los niveles de los complejos E2F4-DP-p107/p130 que se asocian con disminución de los promotores, parece que se sustituye por el E2Fs activador. De manera significativa, la pérdida de los complejos asociados promotor E2F4 se correlaciona con la disociación de los complejos E2F4-DP-proteína y las exportaciones nucleares de los complejos libres E2F4-DP. Por último, in vivo se muestran que los sitios E2F de la B-myb, cdc2 ciclina A2 y los promotores están ocupados principalmente durante G0/G1[240]. Algunos grupos han interpretado esto como evidencia de que estos genes están regulados exclusivamente por la asociación / disociación de los complejos de represión E2F. Sin embargo, como los bajos niveles asociados activan E2Fs, se detectan en la viruta y la pérdida de E2F3 en gran medida perjudica la inducción del ciclo celular dependiente de estos objetivos genes, se concluye que la activación de la transcripción también tiene una función importante en la regulación de estos genes.
Inducción de la salida del ciclo celular y la diferenciación terminal. Análisis de MEFs que se derivan de E2F de proteínas mutantes de ratón han proporcionado una visión bastante buena de las funciones biológicas de la represión de los complejos E2F. MEFs que son mutantes para E2f4 y E2f5 o p107 y p130 tienen defectos en su capacidad para salir del ciclo celular en respuesta a diversas señales de detención del crecimiento, incluyendo la sobreexpresión de p16 y contacto inhibidor[241]. Esto se correlaciona con la expresión inapropiada de un subconjunto de genes de respuesta a E2F[242]. Sin embargo, estas células mutantes todas pueden responder adecuadamente a las señales estimuladoras del crecimiento y no hay ningún cambio detectable en su capacidad proliferativa[243]. Por lo tanto, la pérdida de los complejos de represión E2F-DP afecta sólo la represión de los genes de respuesta a E2F y por lo tanto la capacidad para salir del ciclo celular. Los complejos E2F4/E2F5-DP también parecen ser cruciales para la regulación de la diferenciación. La sobreexpresión de E2F4 es suficiente para la diferenciación de las neuronas[244]. Por otra parte, los defectos del desarrollo en el E2f4, E2f5 y p107; p130 cepas de ratones doble mutantes parecen ser el resultado de la diferenciación celular defectuosa de diversos linajes[245]. Por ejemplo, el defecto primario en el E2f4-/- en ratones es la anemia fetal, que resulta de la maduración aberrante la última etapa de la eritrocitis139. La transferencia adoptiva en los ensayos de progenitoras in vitro muestran que el defecto no está en la producción de las células precursoras, sino en su diferenciación terminal. Por el contrario, la p107-/-, p130-/- en ratones tienen un defecto en el desarrollo de los huesos largos que parece ser el resultado de un condrocito defectuoso162. Los condrocitos no salen del ciclo celular en la etapa de desarrollo correcta, y al someterse a varias rondas de división celular inapropiada antes de que finalmente inicie la diferenciación. Estos estudios establecen un vínculo claro entre el E2Fs represivas y la regulación de la salida del ciclo celular y la diferenciación terminal. Pero todavía hay preguntas que siguen sin resolverse. En primer lugar, ¿cuáles son los papeles relativos de los miembros de la familia pRb en la regulación de la represión? considerando que p107 y p130 se han detectado en asociación con diversos promotores E2F-sensible, pRB es notablemente ausente. Es interesante especular que esto podría implicar la asociación diferencial de las diversas enzimas de modificación de histonas. Por ejemplo, la detención temporal en G0/G1 podría ser mediada a través de la asociación de p107/p130 y sus HDACs asociados, mientras que la liberación permanente del ciclo celular podría ser forzada a través de la unión de la propagación pRB-SUV39H1-HP1 compleja y seguida del efecto silenciador. En segundo lugar, ¿cuál es el papel de los complejos de proteínas E2F represivas en la diferenciación terminal? ¿Son necesarios simplemente para hacer cumplir una detención del ciclo celular que es esencial para la diferenciación, o tienen una función más directa? Varios estudios proporcionan apoyo a este último modelo. Por ejemplo, el CCAAT factores adipogénica / potenciador de las proteínas de unión (C/EBP) y proliferativa peroxisoma activados por los receptores Y (PPAR-Y) se ha demostrado que se unen a E2F y provocan diversas respuestas biológicas[246]. Por otra parte, PRB también interactúa con varios factores de transcripción específicos de tejido, incluyendo C/EBP y un factor de transcripción llamado factor de osteoblastos vinculante básico 1 (Cbfa1[247]). En esta situación, el PRB parece potenciar la actividad transcripcional de estos factores y por lo tanto promover el proceso de diferenciación.
E2F6 es el miembro más recientemente identificado de la familia E2F y sus propiedades biológicas, sólo ahora están comenzando a emerger. Los residuos que son cruciales para la unión al ADN y las actividades de la dimerización E2Fs se conservan en E2F6, pero carece de las secuencias carboxi-terminal que son responsables tanto de la unión a proteínas y la transactivación[248]. Los estudios demostraron que la sobreexpresión E2F6 puede reprimir la respuesta de los genes E2F[249]. Se ha observado que la "represión de dominio" de E2F6 une RING1 y YY1 unión a proteínas (RYBP), un componente de los mamíferos Polycomb (PcG[250]) . En consonancia con esta observación, los socios de E2F6 con numerosas proteínas PcG conocidos in vivo, incluyendo la oncoproteína Bmi-1. Esto sugiere que las propiedades transcripcionalmente represivas de E2F6 están mediados por su capacidad de reclutar el complejo PcG.
Están presentes muchas de las proteínas PcG E2F6-asociadas de control de la expresión del desarrollo de los genes Hox, y en consecuencia la formación de la axial esqueleto[251]. Además, Bmi-1 está implicada en la regulación de la senescencia y tumorigenicidad. De hecho, Bmi-1 fue identificado originalmente como un sitio de inserción común en la leucemia murina, Moloney virus (MoMLV) induce linfomas de células B en ratones transgénicos E-Myc[252]. La actividad transformadora de Bmi-1 parece depender de su capacidad de reprimir la p16INK4a y los genes supresores de tumores p19ARF, que se formulan desde el INK4[253]. En general, se infiere a que Bmi-1 media la represión directa de la transcripción de INK4 a través de su participación en el complejo PcG. Sin embargo, no está claro si el Bmi-1 contiene asociados PcG con este locus. Como p19ARF es un conocido gen E2F de respuesta, es tentador especular que los actos E2F6 como el componente de secuencia específica de unión al ADN de la Bmi-1 contienen complejos PcG. Modelos de ratones mutantes E2f6 serán cruciales en el establecimiento del cómo E2F6 contribuye a la regulación de los patrones de desarrollo y senescencia por el complejo PcG.
Conclusiones parciales. En la actualidad existe considerable evidencia de que en los mamíferos los subgrupos E2F (E2F1, E2F2 y E2F3, E2F4 y E2F5) actúan en la oposición entre sí para mediar la activación o la represión de los genes E2F-respuesta y de ese modo promover ya sea la entrada del ciclo celular o su detención. Los estudios realizados en Drosophila melanogaster brindan gran apoyo a este modelo "push-me-pull-you”. Drosophila tiene sólo dos proteínas E2F: dE2F1, un potente activador transcripcional y dE2F2, que reprime la transcripción de E2F-dependiente[254]. Sorprendentemente, los defectos que resultan de la pérdida dE2f1 son casi completamente suprimidos por la mutación de dE2f2, lo que indica que estas dos proteínas antagonizan entre sí in vivo171. En células de mamíferos, el equilibrio entre la represión y la activación de E2Fs parece ser controlada principalmente por el estado de fosforilación de las proteínas asociadas en respuesta a la detención del crecimiento proliferativo. Sin embargo, dos cuestiones importantes por resolver. En primer lugar, no está claro cómo diferenciar las señales de interfaz con la regulación de estos subgrupos E2F. ¿Utilizan la misma maquinaria del ciclo celular al inhibir la fosforilación de las proteínas de poket y fomentan la salida del ciclo celular, o los cambios adicionales necesarios para cumplir detención permanente del ciclo celular? En segundo lugar, todavía tenemos que entender cómo E2F6 encaja en este cuadro. A falta de datos el circuito no está completo, no está claro si E2F6 regula todos o incluso un subconjunto específico de los genes conocidos E2F-respuesta y si esto sucede en cada ciclo celular o se limita a una fase determinada de desarrollo. Por último, tenemos que establecer cómo influye en la actividad E2F6 y la regulación de los miembros de la familia, y para determinar las consecuencias biológicas de su recurso.
3.20.3 Fase S
3.20.3.1 Replicación de ADN
Naturaleza helicoidal. El documento de investigación biológica más famosos de 1953 - y, posiblemente, de todos los tiempos – fue el de James Watson y Francis Crick donde reportan la descripción de la estructura del ADN[255]. Esta doble hélice de bases apareadas en una estructura, explicó cómo la replicación del ADN podría ocurrir de manera tan precisa y, según los autores, con cautela, declararon: "No escapa a nuestra atención que el apareamiento específico que hemos postulado sugiere inmediatamente un posible mecanismo de copia del material genético". Sin embargo, fue otro documento, también publicado en 1953, pero ahora en gran parte olvidado, que sentó las bases para el modelo del ciclo celular como la conocemos hoy en día.
En 1944 Mac McCarty demostró por primera vez que el ADN es el material de la herencia, la materia llamada de la vida. Hasta entonces, los biólogos pensaban que los genes, las unidades de la herencia, estaban hechas de proteínas[256].
En la década de 1950, la idea de que los cromosomas están hechos de ADN se convino en general. De hecho, Hewson Swift comentó que "el ADN se ha demostrado que poseen interesantes características que han llevado a varios trabajos como para considerarlo un componente esencial de la genética[257]". Sin embargo, el número de moléculas por cromosoma y la naturaleza de su régimen siguió siendo un misterio.
Swift trató esta cuestión en 1950173, con un estudio para calcular la cantidad de ADN por núcleo en los tejidos vegetales. Pregunta si, como se había encontrado a partir de estudios sobre los tejidos de los animales, todas las células vegetales contienen la misma cantidad de ADN (con los gametos contienen la mitad de ese valor), o si no hay patrones cuantitativos tales. Swift hizo mediciones fotométricas en el maíz Feulgen teñido y los núcleos de Tradescantia para demostrar que "el ADN se produce en unidades bien características de la raza o especie". También concluyó que existe una duplicación de este número en la mitosis y una reducción en la meiosis.
Esta duplicación del ADN se estudió con más detalle por Alma Howard y Stephen Pelc en 1953[258]. El uso de 3H-timidina como marcador biológico no se introdujo hasta 1957 (por J. Herbert Taylor y sus colegas[259]), por lo que Howard y Pelc tuvieron que conformar con 32P -una etiqueta insatisfactoria, ya que iluminó todos los compuestos que contienen fósforo-. Por casualidad, sin embargo, los autores encontraron que con la extracción con ácido clorhídrico a 60 °C eliminaban todas las etiquetas de fósforo no-ADN, lo que les permite seguir los cambios en los niveles celulares de ADN. Howard y los estudios de autorradiografía Pelc puso de manifiesto que la síntesis de ADN sólo se produce dentro de un determinado período de tiempo limitado - el medio de la interfase - al principio del proceso de división celular. Este descubrimiento en última instancia condujo a la división del ciclo celular eucariota S, G1, G2, M y fases. El mecanismo detrás de la síntesis de ADN se desenredó cinco años más tarde por Matthew Meselson y Franklin Stahl[260]. Propusieron que una etiqueta radioisotópica que aumenta la densidad del ADN podría permitirles el uso de técnicas de sedimentación para estudiar la distribución del ADN. Para ello, desarrollaron un método para detectar pequeñas diferencias en la densidad de las macromoléculas. Los autores utilizan la sedimentación de gradiente de densidad en seguimiento de la distribución del 15N isótopo pesado del nitrógeno entre las moléculas de ADN después de una 15N-etiquetados en crecimiento exponencial de la bacteria escherichi, fue transferido a un medio de cultivo que contiene el isótopo 14N nitrógeno ordinario.
Meselson y Stahl encontraron que cada molécula de ADN consiste en dos subunidades, dos de los cuales contenían la misma cantidad de nitrógeno. Se descubrió entonces que, una vez después de la adición de 14N, todas las moléculas de ADN contenidas eran la mitad de la cantidad original de la etiqueta, y concluyó que, "Después de la replicación, cada molécula hija ha recibido una subunidad parental". Tomando los dos resultados juntos, se declaró además que "el hecho resulta en una duplicación de replicación molecular".
Meselson y Stahl resumieron sus resultados: "exactamente de acuerdo con las expectativas del modelo de Watson y Crick de la duplicación del ADN", con la advertencia siguiente: "debe hacer hincapié en que no se ha demostrado que las subunidades moleculares que se encuentran en el presente experimento son cadenas de polinucleótidos única o incluso que las moléculas de ADN estudiados corresponden a las moléculas de ADN de una sola propuesta por Watson y Crick". En el transcurso del tiempo se ha demostrado, sin embargo, que sus predicciones resultaron ser correctas.
Origen de replicación. Un origen de replicación es el lugar del cromosoma donde se inicia la replicación de la cadena de ADN. Ya hemos dicho que un principio de la teoría celular, es que todas las células vivas deben replicar su ADN una vez y sólo una vez antes de dividir, y deberán hacerlo con una precisión dura para garantizar que su herencia genética se mantiene intacta. Los estudios clásicos en sistemas bacterianos y virales nos han educado en la idea de que la replicación del ADN consiste en complejos de la ADN polimerasa, que se mantienen bidireccionalmente desde un punto fijo (el origen de replicación). Las células eucariotas poseen normalmente varios cromosomas grandes, lineales, la réplica de la que es probable plantea grandes problemas logísticos adicionales y topológicos, pero el ADN y los componentes de proteína implicada en la determinación de los pasos de la replicación del ADN eucariótico, han sido definidos poco a poco en las últimas tres décadas.
Los primeros trabajos de Stinchcomb, Struhl y Davis, junto con estudios posteriores por Brewer y Fangman y por Huberman y colegas utilizando experimentos plásmidos manejables, los condujo a la identificación de la replicación autónoma (ARS) de secuencias[261]. Estos son elementos cortos de ADN que son capaces de dirigir la replicación de las moléculas de ADN ligadas y que, por tanto, revela las propiedades esperadas de los orígenes de replicación. El pensamiento actual es que el origen de numerosas posiciones a intervalos de aproximadamente 30 kilobases sirven para orquestar la replicación completa del genoma de la levadura. Los orígenes varían en su eficacia y se dividen en clases que son activos en diferentes puntos de la fase S, lo que indica que aún queda mucho por aprender sobre cómo la replicación de los cromosomas eucarióticos se organizan.
La caracterización de los orígenes de replicación de levadura proporcionan los reactivos necesarios para identificar las proteínas celulares que se unen a estos sitios. Un complejo de seis proteínas, conocidas como ORC o complejo de reconocimiento del origen, fue demostrado por Bell y Stillman, al obligar a los orígenes de replicación de la levadura en una forma dependiente de ATP[262], proporcionando una plataforma esencial sobre cómo se basa la replicación del ADN. Diffley Cocker presentó pruebas de que un complejo multiproteico pre-replicativo monta sobre los orígenes de replicación de la levadura en la fase G1 del ciclo[263], y ahora sabemos que esto incluye junto con el ORC genéticamente definidos componentes Cdc6 y el MCM6 (implicadas en el mantenimiento minicromosoma). Este complejo incluye factores de replicación de licencias, que sirven para garantizar que la replicación exista sólo una vez por ciclo celular. El inicio de la replicación es disparado en la fase S por la activación de CDK, después de que las proteínas se asocian para comenzar la fase de elongación de la síntesis de ADN.
Aunque los orígenes de replicación de ADN de mamíferos siguen estando sin concluir, no hay razón para creer que el paradigma de los orígenes pre-programados no será verdad en los eucariontes superiores. Parece probable que los enfoques genómicos y post-genómicos llevarán al progreso en la definición de la ubicación y las propiedades de los orígenes de replicación en el sistema eucariota, y que una mayor caracterización de los componentes proteicos de los aparatos de reproducción, con el tiempo, ofrecerán una imagen más completa de los mecanismos fundamentales que permiten la replicación del ADN precisa y ordenada en organismos eucariotas, actualmente se desarrollan moléculas de ADN circular con un origen de replicación condicional, su procedimiento de preparación y su utilización es vital en terapia génica. La invención de una nueva molécula de ADN con replicación condicional, utilizable en terapia génica o para la producción de proteínas recombinantes ya es una realidad[264].
Para obtener ideas sobre la regulación de los inicios de la replicación, sistemáticamente se asigna replicación origen a lo largo del 1% del genoma humano en células HeLa. Hasta 2008 se han identificado 283 orígenes, 10 veces más de lo que se conocían. Origen es la densidad que se correlaciona fuertemente con los paisajes genómicos, con racimos de pocos espaciados orígenes en las regiones ricas en GC y no orígenes en las grandes regiones GC- pobres. Origen, son secuencias evolutivamente conservadas y la mitad de ellos mapea dentro o cerca de las islas CpG. La mayoría de los orígenes se superponen a elementos reguladores de la transcripción, proporcionando una prueba más de una conexión con la regulación génica. Por otra parte, se identificó c-Jun y c-Fos como importantes reguladores del origen de selección. La mitad de los sitios de inicio de replicación identificados no tienen una configuración de la cromatina abierta, mostrando la ausencia de un vínculo directo con la regulación génica. Mapas de orígenes sugieren que una relativamente estricta programación origen, regula la replicación del genoma humano[265].
Pausa impregnante RAD9. La detención de G2 es seguida del daño en el ADN, por lo que los científicos buscaron en una levadura mutante en ciernes, defectos para esta detención. Al contar el número de brotes presentes en una microcolonia, 10 horas después de la irradiación, los investigadores demostraron que la mayoría de las células fueron detenidas luego de los daños al ADN. Las células que se dividen seguidas a la presencia del daño, una gran proporción de ellas murieron[266]. La fase de una célula se detiene en cuando se encuentra con agentes perjudiciales. Células haploides nativas G1 no tienen cromátida hermana, ni un cromosoma homólogo, por lo que son más sensibles al daño del ADN de la fase S y G2, utilizan la paridad de ADN para reparar roturas de doble cadena. Como tal, estas células G1 detenidas mueren después de la irradiación. Si la función principal de RAD9 es la de mediar esta detención como punto de control, en lugar de reparar el daño en el ADN directamente, RAD9 todavía debe tener la capacidad para reparar - dado el tiempo suficiente.
Cuando las células humanas incurren en daños en el ADN, dos respuestas fundamentales puede seguir, detención del ciclo celular o apoptosis. En humanos RAD9 (hRAD9) la función de p53 está presente en ambos procesos, pero la relación mecanicista entre sus actividades se desconoce; p53 media el control en G1 por la regulación transcripcional de p21. Se ha reportado que hRAD9 y p53, también pueden regular a p21 a nivel transcripcional, se demostró que la sobreexpresión de hRAD9 lleva a un aumento p21 ARN, se demostró que hRAD9 puede controlar la transcripción de genes. Se sugiere que hRAD9 y p53 co-regulan a p21 directamente en la progresión del ciclo celular por mecanismos molecular similares.
hRAD9 se identificó originalmente como un homólogo estructural de la levadura Schizosaccharomyces pombe RAD9 y es capaz de complementar parcialmente la radio sensibilidad, la sensibilidad hidroxiurea y control de defectos de RAD9; además, hRAD9 también funciona como un mediador de muerte celular. El gen supresor tumoral P53 juega un papel fundamental en el control del ciclo celular, la apoptosis y la estabilidad genética[267]. p53 actúa como una secuencia específica de unión al ADN y activa la transcripción por unión de secuencias distintas de ADN. P53 es un regulador negativo de la progresión del ciclo celular y los controles de transición de G1a la fase S del ciclo celular. P53 es necesaria para la respuesta del ciclo celular al daño del ADN causado por la exposición a la radiación[268]. P53 de tipo nativo controla estos procesos mediante el control de la transcripción de genes, mediante su unión a sitios consenso de ADN en las regiones promotoras[269].
P21 es un inhibidor universal de la ciclinas/CDK y funciona como un freno para la progresión del ciclo celular mediante la inhibición de las ciclinas y las quinasas dependientes de ciclinas, como CDK4 y CDK6[270]. P21 es necesario para la detención del ciclo celular mediada por P53, en la radiación inducida para causar daños. Debido a que P21 es un factor clave en el control del ciclo celular, la regulación de P21 es un paso crítico para la respuesta del ciclo celular al daño del ADN y otros tipos de estrés ambiental.
En síntesis, hRAD9 y p53 tienen un papel importante en el control del ciclo celular y la apoptosis, lo que sugiere que estas dos proteínas podrían funcionar coordinadamente. Estos resultados proporcionan evidencia de que hRAD9 y p53 co-regulan a p21 para controlar la progresión del ciclo celular de G1 a S. Usando la tecnología de micromatrices de ADN, se ha demostrado que hRAD9 puede regular la expresión de otros genes, además de p21. Esto sugiere que la modulación de la transcripción génica podría ser un mecanismo por el cual hRAD9 controla múltiples procesos celulares.
E2F y RB. El E2F (un heterodímero de E2F y proteínas DP) de la familia de factores de transcripción, son reguladores importantes de la transición G1 / S del ciclo celular, debido a que E2F puede activar la expresión de los genes en fase S, necesarias para la concesión de replicación de ADN119. También ya hemos dicho que E2F es un represor actuando con un miembro del retinoblastoma (RB). E2F-RB complejos ADN-vinculados reprimen la transcripción al ocluir el dominio de transactivación de E2F y mediante la asociación de la histona deacetilasa (HDAC) que altera la actividad de la estructura de la cromatina[271]. La fosforilación de RB resultante en la transcripción de genes activos E2F impulsa a las células a la fase S119.
Los estudios de células de mamíferos han sugerido que E2F y RB también regulan negativamente la fase S, pero su papel exacto en el control de la replicación del ADN todavía no se ha dilucidado. Del mismo modo, los estados de fosforilación RB son importantes para la finalización de la fase S en forma independiente del gen E2F[272].
3.20.3.2 Reparación de ADN
Un axioma fundamental de la herencia genética es el requisito de estabilidad genética excepcional sobre muchas generaciones de células y organismos. Sin embargo, las células tienen que lidiar con ambientes intra y extracelulares que constantemente desafían la estabilidad química del genoma. Además, aunque la normalidad en las transacciones metabólicas de ADN como la replicación, la recombinación y la reparación son generalmente de gran precisión, hay límites a la fidelidad de estos procesos, dándose el modo de inestabilidad genómica. La evolución darwiniana clásica es, por supuesto, estrictamente dependiente de las muchas fuentes de variabilidad genética que se presentan en las poblaciones de las células germinales, así que la noción de "estabilidad genética excepcional” es relativa.
A pesar de la inestabilidad genética (es decir, las mutaciones) en la línea germinal, se proporciona el escenario en el que la historia de la evolución biológica se desarrolla constantemente, las consecuencias de las mutaciones en las células somáticas - como resultado de daño en el ADN causado por la exposición a agentes ambientales - suelen ser nocivas, como el cáncer en el ser humano. ¿Cómo son las células en los organismos de mamíferos protegidos contra las consecuencias perjudiciales de estas mutaciones? Las respuestas a esta pregunta se derivan en parte de nuestra comprensión del fenómeno de la reparación del ADN en células humanas normales, así como de una enfermedad hereditaria humana llamada xeroderma pigmentosum (XP), que se caracteriza por la reparación del ADN defectuoso y un riesgo notablemente mayor de cáncer de piel que está asociado con la exposición a un carcinógeno ambiental en particular, la radiación ultravioleta.
El daño y la reparación del ADN. Un simple nucleótido (un punto) mutado, suele ser generado por la replicación del ADN que ha adquirido algún tipo de base dañada[273]. El daño de base, a su vez, se deriva de diversos mecanismos intracelulares (daño base espontáneo), así como agentes extracelulares (daño base del medio ambiente) que alteran la química y/o la secuencia de las bases nitrogenadas en ADN[274]. T. Lindahl estima que en conjunto, las diversas fuentes conocidas de daños base espontáneas que es alrededor de 25.000 bases alteradas para el genoma humano por célula al día, de los 3x109 bases en el genoma. Con respecto al daño del ADN del medio ambiente, la radiación ultravioleta es una fuente potente que nos expone en todas partes y el daño del ADN se manifiesta más en la piel. Otra fuente son los carcinógenos químicos, que causan daño al ADN en las células que son accesibles para ellos (sobre todo el tracto aerodigestivo) después de su ingestión, inhalación o su entrada en el cuerpo por medio de otras vías.
Existe la real posibilidad de que las mutaciones causadas por el daño base se realicen mediante la replicación del ADN defectuoso a través de estas lesiones[275]. Pero el daño base también puede dar lugar a la replicación del ADN permanentemente detenido y/o en la transcripción, dando lugar a células muerte[276]. De hecho, la carga de daño base de origen natural y fuentes relacionadas con el ambiente sería incompatible con la vida a menos que las células estén dotadas de mecanismos específicos para la reparación de daños en el ADN y las mutaciones puedan ser mantenidas a niveles razonables. En los últimos años hemos sido testigos de avances impresionantes en nuestra comprensión de los muchos mecanismos por los cuales las células mitigan las consecuencias potencialmente letales y mutagénicas de daño en el ADN. Una de las principales respuestas biológicas al daño del ADN es la reparación del ADN - por vías bioquímicas- por ejemplo, la presencia de uracilo en el ADN y bases mispaired se restauran a su estado nativo193. Por lo tanto, uno puede ver la carga mutacional en un momento dado, tanto en la línea germinal y en células somáticas como el resultado de un equilibrio dinámico entre el grado de daño en el ADN, la eficiencia de reparación del ADN (y otras vías celulares que regulan y modulan la reparación del ADN), y en células en división, la proximidad física de la replicación del ADN dañado avanzan a los sitios por delante del daño.
Reparación por excisión del nucleótido dañado. La reparación de la excisión de nucleótido (NER) es uno de varios mecanismos de reparación del ADN por el cual las bases dañadas se eliminan del genoma. El proceso incluye la extirpación de dichas bases, como parte de un fragmento de oligonucleótidos, a diferencia de la reparación por escisión de bases (BER[277]), por el que el daño o bases inapropiadas son extirpadas como base libre o de reparación de genes (MMR[278]), por la que se extirpa mispaired bases de nucleótidos individuales. Mientras NER en las células humanas es un proceso bioquímico complejo que requiere varias proteínas[279]. Las proteínas humanas necesarias para el NER se montan ordenadas, de forma escalonada en los sitios de daño base que son sustratos de la NER[280] [cxcvii] , [cxcviii] . Este conjunto genera un complejo multiproteico grande (algunas veces referido como la escisión de nucleótidos repairosoma[281]). Los estudios en la levadura, un modelo informativo de muchos aspectos de la NER en humanos, indican que algunos de estos complejos podría ser preensamblados en las células, deliberadamente, no expuestos a daños en el ADN[282]. Sin embargo, las conclusiones de estos estudios han demostrado controversia. El complejo de reparación es una versátil "máquina NER" que pueden reconocer muchos tipos de daño base que a menudo tienen poca o ninguna semejanza estructural o química, una incisión (nick) de ADN a distancias precisas a cada lado del daño base exclusivamente en la dañada cadena de ADN, y fragmentos de oligonucleótidos especiales que incluyen el daño base (ver Figura 3.5). In vitro los tres eventos bioquímicos han sido reproducidos con el uso de proteínas purificadas NER humanos o la levadura.

Fig. 3.6 Las características esenciales de reparación por excisión de nucleótidos
Los primeros pasos de NER en ADN transcripcionalmente en silencio. Los tres eventos que son específicos de NER se entienden con bastante detalle[283]: el reconocimiento del daño, la incisión bimodal de ADN y la extirpación de los fragmentos de oligonucleótidos. La capacidad de la maquinaria NER a reconocer muchos tipos de daño base y discriminar estos del ADN en buen estado, así como el ADN de posibles tipos de daño base que no son sustratos de la NER, ha planteado un reto formidable. Este problema aún no se ha resuelto completamente. Sin embargo, se sabe que varias subunidades de la máquina multiproteica NER se requieren específicamente para este proceso[284]. Estos incluyen una proteína llamada XPC[285], que se encuentra mutado en personas con xeroderma pigmentoso genéticos de complementación GRUPO C (XP-C[286]) , una segunda proteína XP, XPA y un complejo de tres proteínas, conocidas colectivamente como la replicación de la proteína A (RPA[287]). Proteico purificado XPC se une preferentemente a ADN que contiene varios tipos de daño de base que son sustratos de NER (ver Figura 3.5). Sin embargo, los determinantes moleculares precisas que promueven esta unión no se conocen. También hay evidencia de estudios in vitro que la unión de proteínas en el ADN dañado XPC es limitante para el proceso de NER, lo que indica que esta unión es una de las primeras, si no el evento inicial, bioquímicos en NER[288].
El genoma humano contiene dos ortólogos relacionados pero no idénticos del gen RAD23 levadura NER. Las proteínas codificadas por los genes humanos están llamados HHRAD23A y HHRAD23B (por homólogo humano de RAD23[289]). Cuando purificada a partir de células humanas, XPC es un complejo con HHRAD23B[290]. In vitro, la tasa neta de matrícula puede proceder en ausencia de HHRAD23B, pero la eficiencia de la reacción in vitro es mejorado mucho gracias a su presencia. Una tercera proteína llamada centrin2/caltractin1, presente en el centrosoma de varios organismos, ha sido identificada recientemente en el complejo XPC/HHRAD23B y se muestra para ayudar a la estabilización de la proteína por XPC HHRAD23[291]. Esta asociación plantea interesantes posibilidades para las relaciones normativas entre NER y la división celular.
XPA es una metaloproteína que también se une preferentemente a muchos tipos de ADN dañado in vitro y el RPA (antes llamado cadena simple unión a proteínas) se une con avidez a una sola cadena de ADN. Los tipos de daño de base que promueven la NER generando algo de anulación de los dúplex de ADN, promoviendo así la unión de la RPA. XPA y el RPA se cree que se unen al ADN después de la unión de XPC-HHRAD23. Un modelo con el apoyo de experimentos bioquímicos indica que el reconocimiento de todos los tipos de daño base en NER requiere dos elementos fundamentales: la interrupción de la base normal de Watson y Crick, y la química alterada en el apartado de daños, que suele implicar a las bases[292]. Una discusión detallada del reconocimiento de daños de la base durante el NER está fuera del alcance de esta revisión tutorial, sigue siendo un reto importante para explorar.
El reconocimiento específico de los sitios de sustrato para la NER es seguido por el ensamble gradual del resto de la maquinaria de NER, que incluye al menos los polipéptidos que se muestran en la figura 3.5. Entre ellos está uno llamado núcleo subcomplejo factor de transcripción IIH (TFIIH), compuesto por seis subunidades[293].
Estas seis subunidades (junto con las proteínas adicionales) constituyen uno de los varios ARN polimerasa II BASALES factores de transcripción, que son necesarios para la iniciación de la transcripción por la maquinaria de transcripción RNA polimerasa II202. Sigue siendo una pregunta intrigante evolutiva en cuanto a cómo este factor de transcripción basal particular, se convirtió en parte del complejo multiproteico NER. En el centro de este proceso de reparación están las dos helicasas del ADN: XPB y XPD, subunidades de TFIIH, que se cree que favorecen esta función durante la transcripción y el NER[294]. Durante la transcripción, en el proceso de desenrollado (formación de burbujas) facilita la iniciación de nuevos transcriptos de ARN mensajero [ccxvi] . Por el contrario, la corrección de ADN durante la NER por TFIIH genera uniones discretas entre doble cadena y una sola cadena de ADN en los bordes de las estructuras de burbuja[295]. Estas uniones son fundamentales para la incisión correcta de ADN durante la NER.
Otras tres subunidades del complejo NER son integradas por dos endonucleasas que cortan las uniones de doble cadena y una sola cadena de ADN con la polaridad definida. La actividad endonucleasa de XPG corta el ADN dañado cadena 3' a los sitios de base dañados, mientras que la actividad endonucleasa de la proteína ERCC1-heterodimérica XPF corta la cadena dañada 5'. La distancia entre estas incisiones en la cadena de ADN dañado es de 30 nucleótidos. La base dañada siempre se encuentra más cerca de la incisión de 3' que a la incisión de 5'. Sin embargo, la distancia exacta entre las incisiones varía de un tipo de daño base a otro, así como la ubicación exacta de la base dañada con respecto a cada incisión[296]. La presencia de incisiones de ADN complementarias bases dañadas genera un fragmento de oligonucleótidos que se separa del ADN. El mecanismo exacto de esta escisión no se ha establecido. Es casi seguro que no es un proceso pasivo, como base simple de vinculación una distancia de 27-30 nucleótidos se oponen a ello. También es probable que la escisión de oligonucleótidos es temporal, y posiblemente de manera mecánica, ligada a la reparación de síntesis de ADN, de modo que la formación de grandes lagunas de cadena simple que podrían ser susceptibles al ataque de nucleasas, lo que efectivamente corta el genoma, es evitar la excisión durante el oligonucleótido (ver Figura 3.6).
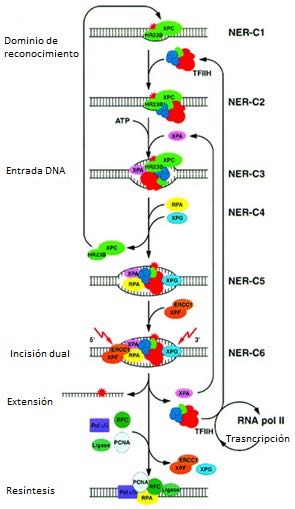
3.20.3.3 GAP 2
Es la tercera fase del ciclo celular, preparación para mitosis o fase M. En G2 se da la ruptura de la membrana nuclear, la condensación de la cromatina y la reorganización del citoesqueleto. Primero precisaremos algunos conceptos. El citoplasma formado por el citosol y citoesqueleto, tiene como función albergar a los orgánulos celulares y contribuir al movimiento de los mismos. Además contiene una compleja red de estructura mecánica de microtúbulos que constituyen el citoesqueleto. El citosol o hialoplasma es el medio acuoso (85% agua) del citoplasma donde ocurren muchos procesos metabólicos que se dan en las células, se divide en ectoplasma (región exterior) y endoplasma (región interna). El citoesqueleto, estructura tridimensional interna de la célula, mantiene a otras estructuras unidas entre sí, y a otras estructuras celulares por diversas proteínas accesorias; además de responder a movimientos mecánicos, da forma a la célula y transportan sustancias.
3.20.3.4 Ruptura de la membrana nuclear
El núcleo separa los contenidos del citoplasma con una estructura de membranas nucleares, donde el núcleo se comporta como un sistema independiente bioquímicamente hablando. La comunicación núcleo y citoplasma es a través del complejo de poro nuclear, controlando selectivamente el paso de RNA’s y proteínas. El núcleo se forma por un sistema de doble membranas concéntricas -interna y exterior-. La membrana nuclear exterior es continua con el E-R. Además, la membrana nuclear exterior es funcionalmente similar a las membranas E-R y delimita la superficie citoplásmica.
La envoltura nuclear consiste en dos membranas recorridas por los complejos de poro nuclear. En la mitosis el poro complejos nuclear se desmantela y se dispersan las membranas. El mecanismo de dispersión es controvertido: un punto de vista es que se alimentan de las membranas en el retículo endoplasmático. Con el uso de extractos de xenopus, los núcleos han sido ensamblados y posteriormente inducidos a la interrupción por la adición de extracto de metafase. Con ayuda de la emisión de campo de microscopía electrónica de barrido se estudia el desmontaje. Sorprendentemente, el retículo endoplasmático, formado por túbulos y membranas de la superficie nuclear, después de la adición de los extractos de la metafase, se observaron vesículas. Los inhibidores de los microtúbulos se desaceleraron, pero no evitaron la remoción de la membrana, mientras que Brefeldin A, que inhibe la formación de vesículas, detiene el desmontaje de membrana, lo que sugiere que es necesaria la vesiculación. Las estructuras que parecían capullos recubiertos observaron brotes, fueron etiquetados como beta-COP. Se ha demostrado que los complejos nucleares del poro se desmantelan y el poro se mantiene cerrado con anterioridad a la ruptura de membrana, lo que sugiere que la ruptura es un proceso activo más que un resultado de la ampliación de los poros nucleares APN. (NE, la envoltura nuclear; el INM, la membrana nuclear interna; ONM, la membrana nuclear externa; NPC, complejo poro nuclear; ER, el retículo endoplásmico). [ccxix] Las membranas nucleares son bicapas fosfolípidas que sólo son permeables a las moléculas no polares pequeñas e impermeables impidiendo el paso a otras moléculas. Las membranas nucleares internas y exteriores se unen en el complejo de poro nuclear [ccxx] . Mirando por debajo de las membranas nucleares encontramos las láminas nucleares, constituidas por proteínas láminas. La lámina nuclear es de unos 10 nm de espesor estructural[297] formadas por una masa nuclear entre los 60 y 80 KDa – kb, kiloDalton-, pertenece a la familia de filamentos intermedios[298]. Se piensa que la lámina nuclear sirve como un sitio de atadura de la cromatina, además de proporcionar el apoyo estructural al núcleo[299]. Las nuevas ideas se apoyan en modelos basados en que los cromosomas y la cromatina crean una interfase en torno al ambiente nuclear que regula la actividad del gen, no sólo durante el ciclo celular, también sirve para producir la estructura funcional que se llama interfase nuclear.
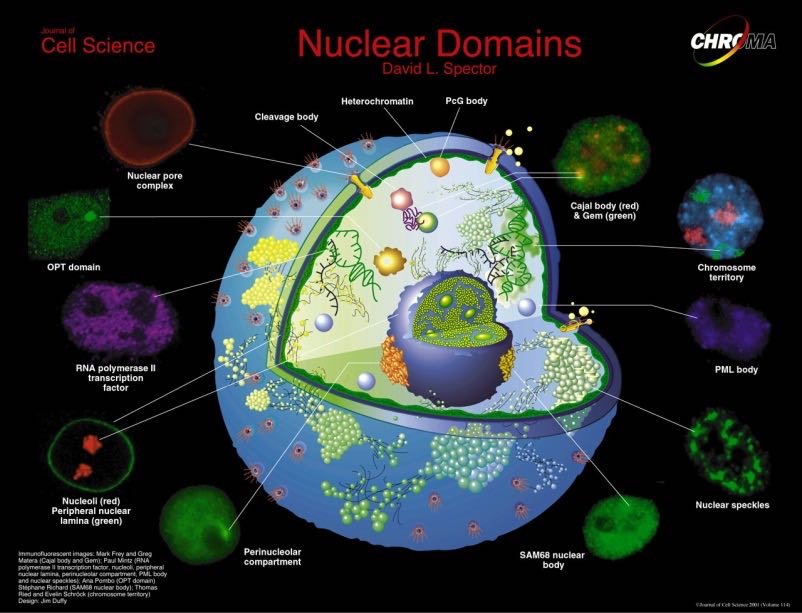
Fig. 3.7. Dominios nucleares[300]
3.20.3.5 Condensación de la cromatina
En 1991, Haaf y Schmid afirman la existencia de un orden de patrones que organizan el núcleo de la célula[301]. Se ha propuesto que repetitivas secuencias actúan como centro estructural de la extensión y condensación de la cromatina[302]. Por lo tanto, la compartimentación de la cromatina podría proporcionar un marco estructural para explicar el eficiente tratamiento de sucesos nucleares[303]. En la base de esta hipótesis, la naturaleza específica de la compartimentación podría reflejar el estado fisiológico de una célula[304]. Sin embargo, cabe destacar que un principio de validez universal del cromosoma no existe.
La idea de la organización territorial de los cromosomas fue primero propuesta por Rabl C. en 1885. Rabl predijo que los cromosomas conservan la integridad estructural, y especula sobre la identidad genética de todo el ciclo celular, postuló que el territorio de cada cromosoma ocuparía una región concreta del núcleo, manteniendo su propia coherencia, y evita mezclarse con los demás territorios.
Ahora, como resultado del desarrollo y los avances técnicos de los métodos in situ, los cromosomas individuales o regiones cromosómicas pueden ser visualizados directamente en el interior del núcleo y sus posiciones pueden ser seguidas en todo el ciclo celular. Por medio de hibridaciones in situ de ADN del cromosoma específico; inmunofluorescencia y microscopía electrónica, es posible su reconstrucción en tres dimensiones, y demás debido a alta resolución en autorradiografía in situ que se han empleado con éxito para estudiar la distribución espacial y funcional de la organización del núcleo en la interfase228. Cremer y colegas en 1993 propusieron un modelo de predicción para la superficie de los territorios cromosómicos y un espacio formado entre ellos proporciona una red similar en tres dimensiones nucleares para la expresión de genes, ARNm y el transporte, llamado el compartimento dominio intercromosómicas (ICD[305]).
3.20.3.6 Reorganización del citoesqueleto
Salida G2 la banda preprofase PPB. El citoesqueleto tiene la apariencia de un gel por su estado más o menos viscoso, su constitución recae básicamente en tres tipos de proteínas: los filamentos de actina, los microtúbulos y los filamentos intermedios. Estos polímeros favorecen el desarrollo de estructuras geométricas perfectas, permiten sufrir cambios: deformaciones y reorganizaciones. El ensamblaje y la reorganización de los filamentos intermedios está ligada a la actina y los microtúbulos a la plectina.
El citoesqueleto tiene un papel fundamental en la mitosis, que es la etapa del ciclo celular en donde los cromosomas duplicados se separan físicamente y forman dos nuevos núcleos, y en la citocinesis durante la partición de una célula en dos. Ambos procesos son llevados a cabo por elementos del citoesqueleto y existen diferencias importantes entre células animales y de plantas. Las plantas no tienen centrosomas y tampoco centriolos. En plantas la estructura del huso mitótico consiste de cientos de microtúbulos y de proteínas que se asocian a éstos.
Citocinesis es el fenómeno por el cual se reorganiza el citoesqueleto. Las células vegetales están rodeadas por muros que definen sus formas y fijan sus posiciones con los tejidos. En consecuencia, el establecimiento de un marco celular de la planta durante el desarrollo depende en gran medida de las posiciones en que nuevos muros se forman durante la citocinesis. Los experimentos con diversos enfoques sobre la base de los estudios clásicos arrojan luz sobre los mecanismos que subyacen el control espacial de la citocinesis.
Hace más de 100 años, los biólogos de plantas reconocieron que la orientación de la división para la mayoría de las células se podría predecir por sus formas. En 1863, Hofmeister observó que las paredes de células nuevas se forman en un plano perpendicular al eje principal de la expansión de células - es decir, perpendicular al eje longitudinal de la célula madre[306]. En 1888, Herrera formuló la regla de que el eje de la división para la mayoría de las células vegetales se corresponde con la trayectoria más corta que reducirá a la mitad el volumen de las células madres. La evidencia experimental apoya la idea de una relación directa entre la forma celular y el plano de la división, que proviene de estudios en los que las formas se han alterado mediante la aplicación de una fuerza de compresión, células esféricas en grupos multicelulares, o aislada por la suspensión de células individuales en medios semi-sólidos[307], se dividen en las orientaciones al azar. Sin embargo, cuando se comprime en formas ovaladas, las células se vuelven muy sesgadas hacia la división en el plano perpendicular, al eje mayor del óvalo.
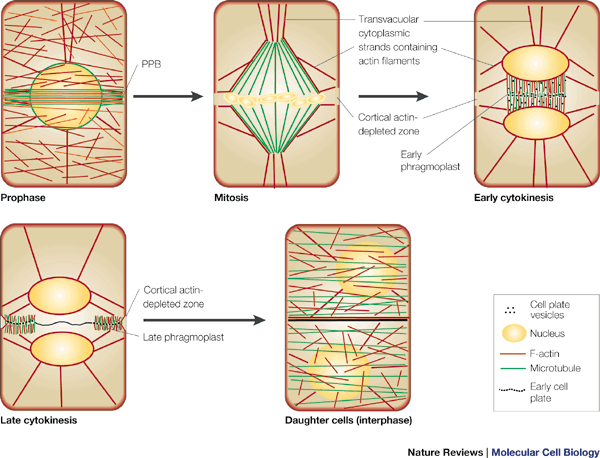
Fig. 3.8. Reorganización del citoesqueleto.
Los filamentos de actina se distribuyen por toda la corteza celular durante la profase, pero algunos están claramente alineados con los microtúbulos del PPB. Cuando los PPB microtúbulos se desmontan a la entrada en mitosis, el componente de la actina del PPB también desaparece, dejando tras de sí una zona de actina empobrecido en la corteza de la célula que persiste y marca el lugar de la división a lo largo de la mitosis y la citocinesis. Después de la terminación de la mitosis, microtúbulos y filamentos de actina se inician entre los núcleos hijos, que guía el movimiento de las vesículas de Golgi-derivados que contienen materiales de la pared celular de la placa celular. A medida que avanza la citocinesis, se expande centrífugamente hasta que se fusiona con la membrana plasmática de los padres y la pared celular en el lugar de la división cortical anteriormente ocupado por PPB.
En el inicio de la mitosis, los microtúbulos corticales en la mayoría de las células vegetales se condensan en un estrecho anillo alrededor del núcleo, la banda preprofase (PPB[308]). Que determina el plano de división celular en el futuro. Junto con los microtúbulos y filamentos citoplásmicos se mueven en el plano de división celular para crear un dominio citoplasma - enriquecido en esta ubicación, la formación de PPB se acompaña de un aumento dramático en la densidad de los microtúbulos perinucelar, que después se reorganizan en el huso mitótico. El PPB en vegetales desaparece por completo durante la cariocinesis, que está mediada por un huso mitótico cuyos polos, en ausencia de centrosomas, no son tan bien enfocados como en las células animales y carece de los microtúbulos astrales. Después de la anafase, se desintegra el huso y una estructura microtubular involucrada en la generación de una pared celular, la placa celular entre los dos núcleos hijos recién formado durante la citocinesis. Inicialmente, se coloca en el centro de la célula en división y tiene la forma de un pequeño cilindro. Sus componentes principales son dos conjuntos opuestos de microtúbulos paralelos que se alinean a lo largo del eje del cilindro y la superposición en un plano central, donde se inicia la formación del chasis de la célula. La formación citoplasmática es lenta durante la citocinesis. En la telofase, la envoltura nuclear se reforma, se descondensan los cromosomas, el huso mitótico se rompe y se forma el fragmoplasto[309].
3.20.3.7 Fase M
3.20.3.7.1 La nanomáquina citoesquelética

Las células eucariontes dependen de los polímeros citoesqueléticos y los motores moleculares para establecer sus formas asimétricas, transportar a los componentes intracelulares y manejar su motilidad. Los biólogos celulares y biofísicos se están acercando experimentalmente a la comprensión de la base molecular del movimiento celular y a explicar qué defectos en las proteínas causan las enfermedades. La mayoría de la maquinaria molecular para motilidad ha evolucionado desde el eucarionte primitivo, comprender un juego limitado de principios generales que puede explicar la motilidad de la mayoría de las células.
Tres polímeros citoesqueléticos[310] - los filamentos de actina (microfilamento), tubulina (microtúbulo) y filamentos intermedios (ver Tabla 1) – participan para mantener la integridad física de células eucariontes; y junto con los motores moleculares, permiten a las células el movimiento. Aunque la motilidad celular como campo de investigación y aplicación en materia de salud es relativamente muy reciente, su empleo se ha extendido como resultado de modelos sobre los mecanismos moleculares, la participación citoesquelética y moléculas de motilidad en muchos aspectos de la función celular, incluso embriología, aprendizaje y memoria, la expansión del cáncer y la patogénesis microbiana. El ensamble cuidadosamente regulado de los polímeros del citoesqueleto y la acción de los motores asociados, es principalmente responsable de establecer la arquitectura celular y así la estructura del tejido.
Tabla 1. Componentes del citoesqueleto
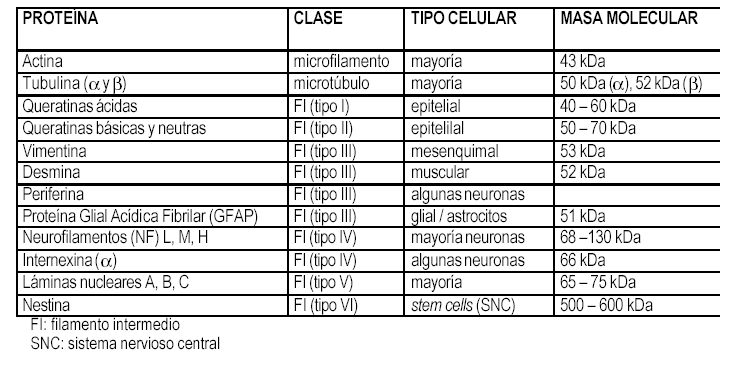
Tabla 2.- Principales fármacos de acción sobre los microfilamentos y microtúbulos.
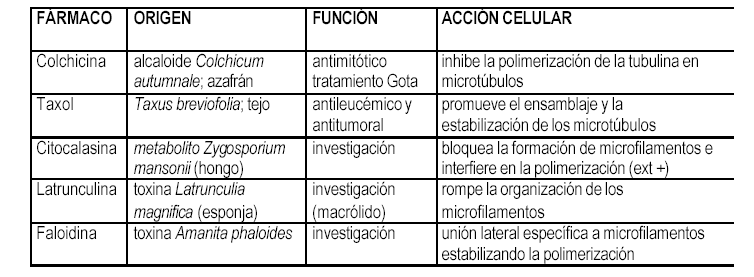
Este apartado pretende introducirnos al fascinante mundo del movimiento mecánico celular. Se explica cómo ensamblaje y desmontaje de microtúbulos transportan algunas moléculas intracelulares, cromosomas y organelos[311]. Las células de locomoción son caracterizadas por una polaridad anterior-posterior externa e interior pronunciada, se cree que esta posición es de importancia estratégica para el movimiento celular direccional y polaridad de la célula[312], además los motores moleculares que actúan recíprocamente con los filamentos actina y microtúbulos para generar la tensión en el citoesqueleto, así como para mover cargas tan grandes como los núcleos y tan pequeñas como las moléculas de ARN[313]. Surgen preguntas del cómo las células usan polímeros citoesqueléticos y motores moleculares para generar la asimetría o, interactividad con la dinámica del aparato de Golgi[314]. Se aprovecha para intentar contestar esta pregunta, que los organismos infecciosos pueden secuestrar el sistema de motilidad para sus propios propósitos[315]; sobre la segregación de cromosomas durante la mitosis y citocinesis[316].
Cuando estudiamos ingeniería observamos una extraordinaria similitud con la geometría de campos magnéticos con limaduras y los polímeros citoesqueléticos, además de los motores que congregan las máquinas complejas temporalmente, para llevar a cabo los procesos vitales con una alta precisión. Las máquinas usadas para la locomoción celular, el transporte intracelular, mitosis y citocinesis consisten en millones de moléculas que se mantienen unidas por relativos enlaces débiles, enlaces no covalentes unen y permite a estas máquinas desmontar y reciclarse.
Las investigaciones citan mutaciones de ankyrin (parte del esqueleto de la membrana) que causa un tipo de arritmia cardíaca,titin en cardiopatías[317] y en myosin-II en los defectos congénitos del cerebro, [ccxlvii] además de otras enfermedades como las de riñón, y diabetes[318].
Sin duda la investigación de la división celular crece porque las posibilidades de aplicación gracias al conocimiento de sus mecanismos pudieran llevar a mejoras en el tratamiento de enfermedades como el cáncer, Alzheimer[319] y por supuesto la nanomáquina que es responsable para el huso mitótico y citocinesis[320]. Pero para entender este camino de enorme esfuerzo y talento hemos agrupado algunos hitos que dieron forma camino a la Biotecnología, ver tabla 3.
Tabla 3. El camino a la Biotecnología.
Decimosexto siglo 1590 El microscopio se inventa por Zacharias Janssen, un fabricante del espectáculo holandés Decimoséptimo siglo 1663 La primeras células son descritas por Robert Hooke 1675 Antony Leeuwenhoek, un relojero holandés, descubre las bacterias. 1796 Científico británico Edward Jenner inocula a un niño para protegerlo de la viruela, el nacimiento de la vacuna de la viruela Decimonoveno siglo 1800 Karl Friedrich Burdach acuña el término “biology” denota el estudio de la morfología humana, la fisiología y psicología. 1833–4 Anselme Payen y Jean-François Persoz aíslan el diastase ( amilasa) en la forma de polvo de la malta cebada y postula la importancia central de enzimas en la biología 1838 Gerardus Johannes Mulder acuña el término “proteína” 1854 Louis Pasteur descubre la fermentación microbiana 1855 Coli Escherichia (E.coli) la bacteria se descubre; ésta se vuelve el caballo de trabajo durante los días modernos de la genética de diseño 1863 Gregor Mendel descubre los rasgos hereditarios más tarde determina “los genes” 1864 Ernst Félix Emmanuel Hoppe-Seyler realiza la primera cristalización de una proteína, la hemoglobina, 1866 Ernst Heinrich Häckel supone que el núcleo de una célula transmite la información hereditaria 1871 Johann Friedrich Miescher aísla una sustancia de los núcleos de las células de la sangre que él llama “nuclein,” qué viene mas tarde a ser llamado ácido nucleico o ADN 1875 Eduard Strasburger describe los procesos mitóticos con precisión en la división celular
|
1877 Wilhelm Friedrich Kühne propone el término “enzima” (significando “levadura”) y distingue las enzimas de los microorganismos que las producen 1878 Carl de Laval inventa la primer centrífuga 1888 Heinrich Wilhelm Gottfried Waldeyer nombra el cromosoma 1880–90 Walther Flemming, Eduard Strasburger, Edouard, Beneden y otros elucidan la esencia de los hechos de división celular y enfatizan la importancia de la igualdad cualitativa y cuantitativa de cromosoma en la distribución en células hijas 1890 Emil Adolfo Von Behring descubre los anticuerpos 1892 Dmitri Iosifovich Ivanovski descubre el virus, agente de enfermedad, más pequeño que las bacterias 1893 Wilhelm Ostwald demuestra que las enzimas son los catalizadores 1897 John Jacob Abel y Alberto C. Crawford aíslan la primer hormona, la epinefrina, después nombrada por Jokichl, Takamine. Vigésimo siglo 1900 Hugo DeVries (Holanda), Jarl Correns (Alemania), y Von de Erich Tschermak-Seysenegg (Austria) demandan haber descubierto independientemente (verificado) Los principios de Gregor Mendel, marcando el principio, de las genéticas modernas 1902 Emil Fischer y Franz Hofmeister demuestran que esas proteínas son los polipéptidos 1903 Carl Neuberg usa por primera vez el término “bioquímica ” 1906 C.W.Woodworth y William Ernest Castle introducen la Drosophila como un nuevo material experimental para los estudios genéticos Mikhail Semenovitch Tsvett usa primero la técnica de cromatografía mientras los pigmentos de la planta se separan de su nombre |
1908 Archibald Edward Garrod reconoce que los productos de un gen son las proteínas 1911 El primer virus causante de cáncer lo descubre Francis Peyton Rous 1912 Alexis Carrel desarrolla la técnica del cultivo de tejido in vitro El Señor William Henry Bragg y Señor William Lawrence Bragg desarrollan la técnica de cristalografía de Radiografía, que se usará después en ella elucidación 3-D de las estructuras de proteínas y los ácidos nucleicos 1913 Alfred Henry Sturtevant desarrolla el primer trazo genético usando las frecuencias crossover como los dimensiones de distancias relativas 1914Se usan bacterias para tratar el alcantarillado la primera vez para en Manchester, Inglaterra 1915 Frederick Twort descubre un virus capaz de infectar y que destruye las bacterias 1917 Félix Hubert D'Herelle, independientemente de Fredelerick Twort, descubre un virus capaz de infectar y destruir bacterias, él llama un bacteriophage 1920 La hormona de crecimiento humana se descubre por Evans 1925 Theodor Svedberg inventa la ultra centrífuga y él determina las proporciones de la sedimentación de proteínas 1928 El señor Alejandro Fleming descubre la acción antibacteriana de la penicilina 1932 M.Kroll y Ernst August Friedrich Ruska construyen el primer microscopio electrónico 1937 George William Marshall Findlay y F.O. MacCullum descubren el interferón 1938 el término “biología molecular” es acuñado |
1941 Selman Abraham Waksman acuña el término “antibiótico” para describir compuestos producidos por los microorganismos, bacteria de muerte. El término “ingeniería genética”es usado por el microbiólogo dinamarqués A.Jost en una conferencia sobre la sexualidad, reproducción en la levadura, en el Instituto Técnico en Lwow, Polonia, 1944 Oswald Avery, Colin MacLeod, y Maclyn McCart demuestran que la tranformación bacterial es causa del ADN 1945 Brand informa del primer análisis del aminoácido completo de una proteína, beta-lactoglobulina, por métodos químicos y microbiológicos. 1946 El primer ejemplo de recombinación genética se registra, combinando el material genético de diferentes virus para formar un nuevo tipo de virus 1947 Bárbara McClintock descubre el elemento transposable o “gen saltador” en maíz. 1948 Benjamín Minge Duggar descubre el aureomicina, el primer antibiótico tetraciclina 1949 Linus Carl con muestras de Pauling muestra las propiedades electroforesis diferentes para hemoglobina normal; esto demuestra que mutaciones genéticas conllevan cambios químico específicos en las moléculas de la proteína. 1950 Inseminación artificial de ganado, se usa el semen helado y es substancialmente cumplido 1952 Alfred Day Hershey y Martha Chase demuestran, en base a su investigación bacteriofago, deducen que el ADN solo lleva la información genética. 1953 James Dewey Watson y Francis Harry Compton Crick describen la estructura molecular del ADN con precisión 1953–4 Vincent du Vigneaud lleva a cabo en laboratorio la primera síntesis de oxitocina de hormonas péptidos y vasopresina 1955 Severo Ochoa y M. Grunberg-Manago descubren la fosforilación del polinucleótido y con éxito sintetizan ARN |
1956 Joe-Hin Tjio y Johan Alberto Levan revisan la estimación de Walther Flemming 1898 de la cuenta de cromosomas humanos de 24 pares, hasta 23 Arthur Kornberg descubre la polimerasa I, de ADN qué lleva a la comprensión de la replicación de ADN 1957 Mahlon Bush Hoagland, Paul Charles Zamecnik, y M.L.Stephenson aíslan el ARNm y postula su función 1958 Francis Harry Compton Crick enuncia el dogma central de la genética molecular, es decir, los flujos de información de ADN a ARN a la proteína 1961 François Jacob y Jacques Lucien postulan la función del mensajero RNA 1965 Genes que llevan la resistencia a los antibióticos en las bacterias se encuentra para residir en los cromosomas en superpequeños llamados “plásmidos” 1966 El código genético se descifra, mientras se demuestra así que una sucesión de tres nucleótidos consistiendo en un codón cada uno determina los 20 aminoácidos 1969 Una enzima se sintetiza por primera vez in vitro 1970 La primera síntesis completa de un gen es cumplida Howard Martin Temin y David Baltimore independientemente descubran los virus: retroviruses—RNA capaz de transcripción inversa, es decir, la síntesis de ADN de una plantilla de ARN La primera enzima de restricción se aísla 1972 La composición de ADN de humanos se descubre, 99% similar al de chimpancés y gorilas. 1973 Stanley Norman Cohen y Herbert Wayne Boyer demuestran que esas enzimas de la restricción pueden usarse para transferir los genes de una especie a otra; Se implantan los plásmidos de ADN con éxito en las células de E.coli, demostrando la posibilidad así de clonar los genes extranjeros en las células bacterianas
|
1975 Una conferencia internacional se emplaza en Asilomar, CA, instando las pautas estrictas para regular la recombinación. La investigación de ADN da los primeros anticuerpos monoclonal. Southern demuestra el eficiente método de hibridación DNA–DNA. 1977 Genentech (San Francisco Sur CA), La primer compañía de la ingeniería genética se funda para usar la recombinación de ADN para producir importantes medicamentos –drogas- Las primeras recombinaciones en el ADN molecular incorporan ADN mamífero, se introduce en las bacterias Se desarrollan los procedimientos para una rápida secuenciación de las secciones largas de ADN –Genoma- La viruela se erradica mundialmente 1978 Somatostatin es producida con el método de recombinación de ADN 1979 La primera hormona de crecimiento humana se sintetiza 1980 La Corte Suprema americana aprueba el principio de las formas de vida genéticamente diseñadas, para patente 1981 La Anemia de la célula “Sickle” se vuelve la primera enfermedad genética en ser diagnosticada directamente a nivel del gen, por análisis de restricción enzimática de ADN El primer animal transgenico, un ratón 1982 Un nuevo síndrome severo es caracterizado, el deterioro del sistema inmunológico, se reconoce y dado el nombre de Síndrome de inmunodeficiencia Adquirida (AIDS más conocido como SIDA) La primera proteína genéticamente diseñada, Humulin ®, es aceptada para el tratamiento de diabetes 1983 Kary B. Mullis inventa un método, la reacción en cadena de la polimerasa (PCR), un método por clonar rápida y fácilmente El primer cromosoma artificial se sintetiza 1984 El genoma entero del virus de HIV se clona y se secuencia.
|
1985 Tomando las huellas dactilares genético se introduce en las salas del tribunal americano. Las plantas genéticamente diseñadas resistente a los insectos, los virus se prueban en campo . 1987 La primera prueba en campo de una bacteria genéticamente alterada que inhibe la formación de escarcha en la cosecha de las plantas de fresa y patata en California 1988 Congreso aprueba el fondo del Proyecto Genoma. 1989 El gen responsable para la fibrosis cística es descubierto 1990 Un esfuerzo internacional por trazar todos los genes en el cuerpo humano se lanza bajo el patrocinio del Proyecto de Genoma Humano. El primer gen experimental federalmente aprobado para el tratamiento de la terapia es un asunto humano con éxito realizado La primera vaca de lechería transgénica se crea 1994 El primer gen de cáncer de pecho se descubre 1995 El primer trasplante de médula de hueso mandril-humano se realiza en un paciente con SIDA 1996 Un gen asociado con la enfermedad de Parkinson se descubre 1997 Es clonando un animal, una oveja, se informa por científicos escocés
|
1998 La secuencia del genoma de un animal, -C. elegans, es completada Primer cultivo in vitro células madre embrionarias humanas 1999 La secuencia del genoma de Drosophila se completa Vigésimo primer siglo 2000 Se producen cerdos clonados de las células de cerdo. El primer proyecto de la sucesión del genoma humano es completado 2001 El Proyecto de Genoma Humano estima que el genoma contiene aproximadamente 30,000 genes proteína-codificada. 5 de diciembre del 2002 Inicia el comparativo del genoma del ratón y el humano. 25 Abril 2002
Tecnología de Microarreglos de ADN que pueden descubrir la presencia o la expresión de miles de genes simultáneamente son una herramienta importante en la interpretación de la masa de información genética que sale de los programas de secuenciación del genoma.
|
25 Julio 2002
Proteger el vasto número de compuestos químicos para hallar un poco que pueda volverse las drogas del futuro, es la tarea de los laboratorios clínicos del siglo XXI, esta tecnología desafía el ensayo, muchos ensayos, gran velocidad y bajo costo. [cclvi] 24 de Abril 2003 Datos útiles extraídos de la sucesión del genoma humano es un desafío mayor pero Lisa Melton examina los pasos hacia el genotipo personal. [cclvii] 17 de julio del 2003 Una nueva escala de estudios de expresión de genes en los gusanos nos permite vislumbrar la bioquímica compleja el rango de vida - lifespan-. [cclviii] 8 de julio del 2010 VRC01 en contacto con gp120 principalmente, a través de las regiones V - derivados de genes alterados sustancialmente a partir de sus precursores genómicos, facilita la neutralización del VIH -1 por anticuerpos humanos naturales [cclix] |
3.20.3.7.2 Mitosis
La mitosis es una serie ordenada de eventos fundamentalmente mecánicos en que se mueven copias idénticas del genoma a dos lugares discretos dentro de la célula en división (ver figura 3.10). Las vías para coordinar los microtúbulos base-MT (huso mitótico) y base-Actina (anillo contráctil) durante la mitosis y citocinesis, ya la investigación científica las revela y en las ciencias de la salud aplica este conocimiento para combatir las enfermedades.
En la división celular, la estructura citoesquelética formada por microtúbulos organiza dentro de un huso bipolar los cromosomas, polarización que segrega los cromosomas reproducidos en dos células. La exactitud de la segregación se basa en la atadura de todos los cromosomas antes de que la anafase inicie, en la atadura de la segregación ocurren dos mecanismos, uno de búsqueda y otro de captura al encoger y crecer los microtúbulos que emanan del centrómero, éste último actúa como separadores recíprocamente con los cromosomas condensados[321]. Algunos microtúbulos alcanzan una región llamada proteinaceous especializada y localizada en el centrómero al azar (cinetocoro, Fibra-K), se reclutan progresivamente otros microtúbulos fijando el cromosoma eficazmente al polo del huso. La acción similar entre la Fibra-K hermana y el polo opuesto lleva a la biorientación del cromosoma con microtúbulos que no participan en formación de K-Fibras o actúan recíprocamente como brazos del cromosoma. Cuando todos los eventos tienen lugar cada cromosoma de la célula en división se sujeta simultáneamente a fuerzas polares que actúan en la cinetocoros con la fuerza antipolo[322]. La combinación de estos resultados de fuerzas en movimiento de los cromosomas, contribuye su biorientación y su alineación en la metafase. En la mayoría de las células, una vía favorablemente de transducción señalada, llamada ‘punto de chequeo' del huso, supervisa estos eventos e impide a las células entrar en la anafase, y a los cromosomas segregar y emigrar hacia los polos opuestos prematuramente[323].
El estudio de la Mitosis data de 1880[324], (ver tabla 3), durante este tiempo las especulaciones sobre las fuerzas polares de eyección han planteado: son generadas simplemente por el impacto del crecimiento de microtúbulos en los cromosomas, los microtúbulos amitóticos son muy inestables y la cromatina mitótica es muy elástica, apoyados en investigación con micropipetas. Otra posibilidad que ha sido considerada y ahora muy aceptada, es que los motores moleculares que se localizan a lo largo de los brazos del cromosoma podrían generar estas fuerzas. Durante los últimos años, esta idea se ha reforzado con la identificación de algunas proteínas como kinesin (KLPs) asociadas con los brazos del cromosoma. Iniciando el siglo XXI un buen candidato para un motor de eyección polar fue identificado. Este motor es Xenopus (human Kid, kinesin-like DNA binding protein) asociado con los cromosomas durante la mitosis[325].
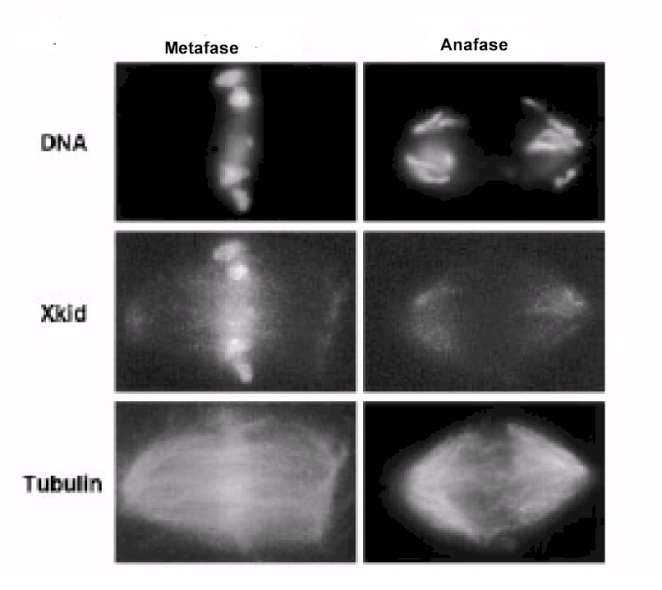
Fig. 3.9. Fuerzas de eyección polares.

Fig. 3.10. Eventos Mitóticos
En figura 3.9, Xkid no se requiere para la reunión del huso pero es esencial para la alineación del cromosoma, mueve los brazos de los cromosomas hacia el ecuador del huso, empezando en las fases tempranas de la mitosis y continuando hasta metafase. Juega un papel en la alineación del cromosoma, Xkid debe degradarse a través de una senda ubiquitin-dependiente para permitir la migración de cromátidos a los polos durante la anafase.
3.20.8 Motores moleculares
Debido a sus propiedades intrínsecas, los motores moleculares han sido considerados los candidatos ideales por generar las fuerzas involucradas en los movimientos del cromosoma durante la mitosis. De hecho, durante los últimos años, se han encontrado varios motores moleculares que localizan a los cromosomas y median las clases diferentes de interacciones cromosoma–microtúbulo que se perfiló anteriormente[326].
En la luz de recientes resultados,269 nosotros repasamos el conocimiento actual de los papeles jugados por los motores cromosoma-asociados durante la mitosis. Interesantemente, algunos de ellos no funcionan convencionalmente, es decir, por la carga de tranlocación a lo largo del microtúbulo, pero los papeles relacionaron a la atadura del microtúbulo, reunión del huso y la actividad de punto de control de huso.
Las fluctuaciones de los motores actina cerebral[327] y cromatina-asociados llevan a la alineación del cromosoma a la metafase. Las fluctuaciones No-equilibradas abstractas como éstas, pueden manejar el transporte vectorial a lo largo de una estructura anisótropa en un medio isotermal torciendo el efecto de ruido termal (kBT). Se llaman a menudo a estos mecanismos basados en este principio, mecanismo Browniano ratchet y se han invocado como una posible explicación para el funcionamiento de motores biomoleculares y bombas[328]. No pretendiendo hacer una profunda discusión termodinámica y cinética para el funcionamiento Browniano ratchet microscópico, podemos asegurar que la mitosis bajo las condiciones pertinente a la biología es un movimiento Browniano[329]. Además Winds sugiere que bombas moleculares como las Na, K-ATPasa y los motores moleculares como la quinesina –kinesin[330]- y miosina –myosin[331]- puedan compartir un mecanismo subyacente común. Timothy C. Lestón, explica la correlación en la flexibilidad de movimientos de proteínas a lo largo de una exhaustiva vida en la investigación científica en este importante campo, además da algunos ejemplos de motores moleculares, se recomienda apreciable lector, revisar este fascinante trabajo[332]-.
Las fuerzas de eyección polares son investigadas en laboratorios altamente tecnificados, se generan por microtúbulos que actúan recíprocamente con los brazos del cromosoma (ver Imag3 microscópicas 3D en http://www.wadsworth.org/databank/cells.htm[333]). La proporción de estos microtúbulos es inconstante entre las especies y cosas correlativas con la eficacia de alineación del cromosoma y estabilización en el plano metafase[334]. Esta observación apoya la idea de que la fuerza de eyección polar juega un papel importante en la alineación del cromosoma en el plano metafase y errores a partir de puntos de control generan enfermedades como el cáncer[335].
Punto de control del huso, supervisa el cumplimiento de requisitos específicos antes de que la célula pueda entrar en la anafase. Estos requisitos incluyen la atadura de microtúbulos a cada cinetocoro, la formación de las K-fibras y el establecimiento de tensión entre el cinetocoro de los defectos en cualquiera de estos eventos previenen el punto de control del huso de volverse inactivos y así impiden a las células entrar en la anafase.
Los recientes datos sugieren que algunos motores del cinetocoro pueden cumplir las funciones de punto de control. La microinyección celular de anticuerpos dirigidos contra el dominio CENP-E no daña la alineación del cromosoma pero inhibe la transición a la anafase, implicando que además de su función estabilizadora las interacciones del cinetocoro–microtubular, “CENP-E” está envuelto en el mando de abordaje de la anafase. Este freno requiere BUBR1, un componente de la maquinaria del punto de control. CENP-E actúa recíproca y directamente con BUBR1 – BUB1 un gen de levadura exigido para asegurar la progresión correcta de la reunión del huso mitótico, son dos las proteínas quinasas relacionadas en humanos BUB1 y BUBR1. Se localizan en el cinetocoro durante la anafase, en las cuales se especula el papel de retardo de la anafase hasta que todos los cromosomas logren la atadura correcta del huso bipolar- pero no se requiere para la localización del cinetocoro de BUBR1 o Mad2, otro componente de punto de control de huso esencial.[336] Así, CENP-E no se requiere para la activación de punto de control de huso y probablemente actúa bajo el flujo de la maquinaria de punto de control de huso, bajo el mando de BUBR1, y puede exigir imponer el punto de control del huso en alto. Es posible que, en estos extractos, CENP-E cumple funciones de punto de control diferentes que se llevan a cabo por motores distintos en las células somáticas. La reciente identificación de CENP-E en Drosophila presta el apoyo a esta idea[337].
Se pensaba inicialmente que Dynein, era el primer motor en ser encontrado en cinetocoro, era un primer candidato para las fuerzas polares generadoras que mueve el microtúbulo hacia los extremos enfocados a los polos del huso. De hecho, Dynein puede controlar el movimiento de cromosoma en la corriente dominante que ocurre inmediatamente después de la primera interacción entre un microtubulo y un cinetocoro. [ccxcv] La importancia de este evento es el papel extenso que este motor juega en la alineación del cromosoma en plano metafase, sin embargo, incierto. De hecho, ni la inhibición de “Rough Deal” (Vara) ni de Z-blanco 10 (Zw10), las proteínas conservadas que Dynein designa al cinetocoro daña la alineación del cromosoma[338]. Dynein parece ser principalmente activa durante la anafase en el punto que realmente se requiere para el movimiento del cromosoma a los polos[339].
La exactitud de la segregación de los cromosomas descansa en la eficiencia de los motores moleculares asociados. Las fuerzas ejercidas sobre los cromosomas y las moléculas importantes ya descritas en líneas atrás. Dynein y Xkid son los únicos motores en actividad convencional, manejando cromosomas como actividad convencional, es decir, manejando cromosomas como carga a lo largo del microtúbulo. Alternativamente, los movimientos durante la mitosis pueden ser principalmente impulsados por la dinámica microtubular, que parece ser regulada por los motores ya identificados. En el cinetocoro, CENP-E y Xkcm1/MCAK pueden funcionar cooperativamente. Es factible que estas actividades combinadas puedan ser suficientes para generar una corriente polar de movimiento durante la mitosis.
Aunque especulativo a estas alturas del desarrollo científico, podríamos pensar que un mecanismo similar podría permanecer oculto a las herramientas tecnológicas del laboratorio del umbral del siglo XXI, a nivel de los brazos del cromosoma. Motores moleculares acoplados a factores que promueven el crecimiento microtubular podrían estar contribuyendo a las fuerzas de eyección polar y migración de cromosomas hacia el plano metafase. Un trabajo con una mayor integración multidisciplinar, requiere la caracterización de todos los motores interactuando y los modos de regulación que permiten los movimientos coordinados de los cromosomas a lo largo del curso de la mitosis.
Los hechos expuestos en los reportes de investigación ya referidos mantienen la idea de que motores cinetocoros descansan en un aspecto mecánico del control del huso, esto abre nuevas líneas de investigación en el campo de motores moleculares.
Los puntos de control y señalización, el objetivo y el estímulo de actividad ya son en alguna manera identificados. Los motores moleculares están emergiendo como el eslabón perdido entre los estímulos y las vías de señalización. Sin embargo, los motores involucrados y sus modos exactos de acción, actividad y la imposición de silencio al punto de control del huso todavía tienen que ser caracterizados totalmente. Como conclusión nos surgen muchas preguntas irresolutas acerca de las contribuciones respectivas de tensión y atadura de Fibras-K en la actividad de punto de control. Sin duda un aspecto orientador es el hecho de que los motores cinetocoros moleculares son importantes para el extenso y complejo proceso mitótico.
El desarrollo del huso mitótico y el anillo contráctil, la “selección natural” ha diseñado con precisión fascinante, movimientos mecánicos generados por un conjunto de proteínas citoesqueléticas. Estas máquinas ya se mencionó que segregan los cromosomas y dividen la célula con una enorme exactitud. También mencionamos como corrientes de investigación científicas actuales se centran en los mecanismos, regulación morfogénesis del huso, motilidad de cromosomas y la citocinesis, dando énfasis en cómo los dinámicos conjuntos de polímeros del citoesqueleto y los motores moleculares cooperan para generar las fuerzas que guían la célula a través de la mitosis y citocinesis.
Durante el siglo décimo, el descubrimiento de la reproducción celular, por división, iluminaron el mismo origen celular y se volvieron estos conocimientos; la piedra angular de la teoría celular. En este escenario aparecen los esfingolípidos, son componentes ubicuos de membranas eucariontes y se requieren particularmente en la membrana del plasma. Clásicamente se ha considerado juegan un papel estructural en las membranas, para la vía metabólica, además, se reconoce ahora como un sistema de señalización importante. Metabolitos derivados del rompimiento del complejo esfingolípido o algunas veces por la síntesis novo, son favorablemente moléculas bioactivas que se implican como segundos mensajeros que median diversas funciones celulares. El metabolito más estudiado es la ceramidasa, un componente central de la vía esfingolípida. Ceramidasa no sólo es un ladrillo para la síntesis del esfingolípido, se cita además un papel de mensajero en stress en muchos sistemas biológicos, incluso en respuesta al golpe de calor o vías apoptosis, senescencia celular, el ciclo celular y diferenciación[340]. Ver MCRI https://www.tuftsmedicalcenter.org/research-clinical-trials/institutes-centers-labs/molecular-cardiology-research-institute/publications)
Referencias
[1] Grzybowski, Andrzej & Sak, Jaroslaw & Pawlikowski, Jakub. (2012). The history of scientific concepts of vision in relation to Ludwik Fleck’s thought-styles. Acta ophthalmologica. 91. 10.1111/j.1755-3768.2012.02450.x.
[2] Neidhoefer, Claudio. (2021). On the Evolution of the Biological Framework for Insight. Philosophies. 6. 43. 10.3390/philosophies6020043.
[3] Jiang, Ching-Fen & Hsu, Shan-hui & Tsai, Ka-Pei & Tsai, Ming-Hong. (2015). Segmentation and tracking of stem cells in time lapse microscopy to quantify dynamic behavioral changes during spheroid formation: Stem Cell Tracking to Quantify Dynamic Behaviors. Cytometry. Part A : the journal of the International Society for Analytical Cytology. 87. 10.1002/cyto.a.22642.
[4] Grew N (1682) The anatomy of plants with an idea of a philosophical history of plants, and several
other lectures read before the royal society. Rawlins W, London
[5] Saks, Valdur & Beraud, Nathalie & Wallimann, Theo. (2008). Metabolic Compartmentation – A System Level Property of Muscle Cells: Real Problems of Diffusion in Living Cells. International Journal of Molecular Sciences. 9. 751-67. 10.3390/ijms9050751.
[6] Brown R (1833) On the organs and mode of fecundation in Orchideae and Asclepiadeae. Trans
Linn Soc Lond 16:685–745
[7] Schwann, Theodore. (1993). Microscopial Researches into the Accordance in the Structure and Growth of Animals and Plants. Obesity research. 1. 408-18. 10.1002/j.1550-8528.1993.tb00021.x.
[8] BAKER, J. (1948). The Cell-Theory: a Restatement, History, and Critique. The Quarterly journal of microscopical science. 89. 103-25.
[9] Benda, C.. (2021). Ueber die Spermatogenese der Vertebraten und höherer Evertebraten, II. Theil: Die Histiogenese der Spermien. Arch Anat Physiol. 73. 393-398.
[10] Sagan, L. (1993). The origin of mitosing cells. The Journal of NIH research : life sciences research and news about the National Institutes of Health and the Alcohol, Drug Abuse, and Mental Health Administration. 5. 65-72.
[11] F, J.. (2021). Die Reizleitung und die reizleitenden Strukturen bei den Pflanzen. Nature. 64. 10.1038/064371a0.
[12] van Bel A (2018) Plasmodesmata: a history of conceptual surprises. In: Balus
Concepts in cell biology – history and evolution, Plant cell monographs. Springer, Heidelberg
[13] Raghavendra, Agepati & Sane, Prafullachandra & Mohanty, Prasanna. (2003). Photosynthesis research in India: Transition from yield physiology into molecular biology. Photosynthesis research. 76. 435-50. 10.1023/A:1024934432008.
[14] Martin, Cyrus. (2017). Wilhelm Hofmeister and the foundations of plant science. Current Biology. 27. R853-R855. 10.1016/j.cub.2017.08.039.
[15] Ford, Brian. (2009). Did Physics Matter to the Pioneers of Microscopy?. Advances in Imaging and Electron Physics - ADV IMAG ELECTRON PHYS. 158. 27-87. 10.1016/S1076-5670(09)00007
[16] Leskova, Alexandra & Kusá, Zuzana & Labajová, Mária & Krausko, Miroslav & Jasik, Jan. (2019). The Photoconvertible Fluorescent Protein Dendra2 Tag as a Tool to Investigate Intracellular Protein Dynamics. 10.1007/978-1-4939-9469-4_13.
[17] Volkmann, Dieter & Baluska, Frantisek & Menzel, Diedrik. (2012). Eduard Strasburger (1844-1912): Founder of modern plant cell biology. Protoplasma. 249. 1163-72. 10.1007/s00709-012-0406-6.
[18] Guttenberg, Hermann. (1943). Kulturversuche mit isolierten Pflanzenzellen. Planta. 33. 576-588. 10.1007/BF01916543.
[19] Twaij, Baan & Jazar, Zena & Hasan, Md. (2020). Trends in the use of tissue culture, applications and future aspects. International Journal of Plant Biology. 11. 10.4081/pb.2020.8385.
[20] Klíma, Petr & ?ermák, Vojt?ch & Srba, Miroslav & Muller, Karel & Petrášek, Jan & Šonka, Josef & Fischer, Lukas & Opatrný, Zden?k. (2019). Plant Cell Lines in Cell Morphogenesis Research: From Phenotyping to -Omics. 10.1007/978-1-4939-9469-4_25.
[21] Vasil IK (2008) A history of plant biotechnology: from the cell theory of Schleiden and Schwann
to biotech crops. Plant Cell Rep 27:1423–1440
[22] Casadevall, Arturo & Fang, Ferric. (2016). Revolutionary Science. mBio. 7. e00158-16. 10.1128/mBio.00158-16.
[23] Karch, Christopher & Burkhard, Peter. (2016). Vaccine Technologies: From Whole Organisms to Rationally Designed Protein Assemblies. Biochemical Pharmacology. 120. 10.1016/j.bcp.2016.05.00
[24] Trewavas, Tony. (2016). Plant Intelligence: An Overview. BioScience. 66. biw048. 10.1093/biosci/biw048.
[25] Wilkins, Adam. (2012). Evolution: A View from the 21st Century. Genome biology and evolution. 4. 10.1093/gbe/evs008.
[26] Maienschein, Jane. (2020). Cell Theory and Development. 10.1201/9781003070818-29.
[27] Miguel, Virginia & Villarreal, Marcos & García, Daniel. (2019). Effects of gabergic phenols on the dynamic and structure of lipid bilayers: A molecular dynamic simulation approach. PLOS ONE. 14. e0218042. 10.1371/journal.pone.0218042.
[28] Lombard, Jonathan. (2014). Once upon a time the cell membranes: 175 years of cell boundary research. Biology direct. 9. 32. 10.1186/s13062-014-0032-7.
[29] Sahi, Vaidurya & Wadekar, Hanumant & Ravi, Nagganatha & Thangavelu, Arumugam & Morita, Eugene & Abe, Shunnosuke. (2012). A molecular insight into Darwin’s “plant brain hypothesis” through expression pattern study of the MKRN gene in plant embryo compared with mouse embryo. Plant signaling & behavior. 7. 375-81. 10.4161/psb.19094.
[30] Journal of Theoretical Biology - J THEOR BIOL. 103. 645-648. 10.1016/0022-5193(83)90287-4.
[31] Sekereš, Juraj & Zárský, Viktor. (2018). 180 Years of the Cell: From Matthias Jakob Schleiden to the Cell Biology of the Twenty-First Century. 10.1007/978-3-319-69944-8_2.
[32] Roth, Siegfried. (2011). Mathematics and biology: A Kantian view on the history of pattern formation theory. Development genes and evolution. 221. 255-79. 10.1007/s00427-011-0378-0.
[33] Harris H (2000) The birth of the cell. Yale University Press, New Haven. isbn:0-300-08295-9
[34] Wainer,. (2019). Knowledge Production and Knowledge of Life. CR: The New Centennial Review. 19. 129. 10.14321/crnewcentrevi.19.3.0129.
[35] Welch, G. & Clegg, James. (2012). Reply to “Letter to the editor:'Systemic cell theory, protoplasmic theory, and their logic of explanation'”. AJP -Cell Physiology on Mayphysiology.org Downloaded from. 29. 10.1152/ajpcell.00159.2010.
[36] Sekereš, Juraj & Zárský, Viktor. (2018). 180 Years of the Cell: From Matthias Jakob Schleiden to the Cell Biology of the Twenty-First Century. 10.1007/978-3-319-69944-8_2.
[37] Gupta, Ritika & Mitra, Saheli & Chowdhury, Subhadip & Das, Gangadhar & Priyadarshini, Richa & Mukhopadhyay, Mrinmay & Ghosh, Sajal. (2021). Discerning perturbed assembly of lipids in a model membrane in presence of violacein. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1863. 183647. 10.1016/j.bbamem.2021.183647.
[38] Fazekas, Tamás. (2021). Jan Evangelista Purkinje (1797-1869). Kaleidoscope history. 11. 81-85. 10.17107/KH.2021.22.81-95.
[39] Regöly-Mérey, G. (1968). [The life and philosophy of Johannes Müller].. Orvosi hetilap. 109. 1664-6.
[40] Sazer, Shelley & Schiessel, Helmut. (2017). The Biology and Polymer Physics Underlying Large Scale Chromosome Organization. Traffic. 19. 10.1111/tra.12539.
[41] Gans, Carl. (1983). Book Review:Natural History of Amphibians and Reptiles in Wisconsin. Richard Carl Vogt. Quarterly Review of Biology - QUART REV BIOL. 58. 10.1086/413132.
[42] McIntosh, Richard. (2021). Seminars in cell and developmental biology: Anaphase A. Seminars in Cell & Developmental Biology. 10.1016/j.semcdb.2021.03.009.
[43] Junquera, Santiago. (2010). Ramón y Cajal, microbiologist. International Microbiology; Vol. 3, Núm. 1 (2000); 59-61.
[44] Liu D (2016) The cell and protoplasm as container, object, and substance, 1835–1861. J Hist Biol.
doi:https://doi.org/10.1007/s10739-016-9460-9
[45] Saguchi, S.. (1954). On the fundamental structure of protoplasm. Protoplasma. 43. 262-277. 10.1007/BF01250991
[46] Wilson EB (1899) The structure of protoplasm. Science 10:33–45
[47] Schuldiner, Maya & Schwappach, Blanche. (2013). From rags to riches — The history of the endoplasmic reticulum. Biochimica et biophysica acta. 1833. 10.1016/j.bbamcr.2013.03.005.
[48] Kutschera, Ulrich & Niklas, Karl. (2005). Endosymbiosis, cell evolution, and speciation. Theory in biosciences = Theorie in den Biowissenschaften. 124. 1-24. 10.1016/j.thbio.2005.04.001.
[49] Mcfadden, Geoffrey. (2001). Primary and secondary endosymbiosis and the origin of plastids. Journal of Phycology - J PHYCOL. 37. 951-959. 10.1046/j.1529-8817.2001.01126.x.
[50] Yang, Yue & He, Ping-Ya & Zhang, Yi & Li, Ning. (2020). Natural Products Targeting the Mitochondria in Cancers. Molecules. 26. 92. 10.3390/molecules26010092.
[51] Dingjan, Tamir & Futerman, Anthony. (2021). The fine-tuning of cell membrane lipid bilayers accentuates their compositional complexity. BioEssays. 43. 10.1002/bies.2021000
[52] Richmond, Marsha. (2000). T.H. Huxley's Criticism of German Cell Theory: An Epigenetic and Physiological Interpretation of Cell Structure. Journal of the history of biology. 33. 247-89. 10.1023/A:1004881730937.
[53] Waisse, Silvia & Alfonso-Goldfarb, Ana. (2009). Mathematics Ab Ovo: Hans Driesch and Entwicklungsmechanik. History and philosophy of the life sciences. 31. 35-54.
[54] Sander, Klaus. (1997). Landmarks in Developmental Biology 1883–1924. 10.1007/978-3-642-60492-8.
[55] Borrello, Mark. (2003). [Book Review: Readers of the Book of Life: Contextualizing Developmental Evolutionary Biology]. Quarterly Review of Biology - QUART REV BIOL. 78. 461-462. 10.1086/382371.
[56] Ratcliff, William & Denison, R & Borrello, Mark & Travisano, Michael. (2012). Experimental evolution of multicellularity. Proceedings of the National Academy of Sciences of the United States of America. 109. 1595-600. 10.1073/pnas.1115323109.
[57] OPPENHEIMER, JANE. (1966). The Growth and Development of Developmental Biology. 10.1016/B978-0-12-395618-7.50005-6.
[58] Gilbert SF, Opitz JM, Raff RA (1996) Resynthesizing evolutionary and developmental biology.
Dev Biol 173(2):357–372
[59] Vienne, Florence. (2017). Worlds Conflicting: The Cell Theories of François-Vincent Raspail and Theodor Schwann. Historical Studies in the Natural Sciences. 47. 629-652. 10.1525/hsns.2017.47.5.62
[60] Breindel, Leonard & Yu, Jianchao & Burz, David & Shekhtman, Alexander. (2020). Intact ribosomes drive the formation of protein quinary structure. PLOS ONE. 15. e0232015. 10.1371/journal.pone.0232015.
[61] Quastel, J.H.. (1984). The development of biochemistry in the 20th century. Canadian journal of biochemistry and cell biology = Revue canadienne de biochimie et biologie cellulaire. 62. 1103-10. 10.1139/o84-144.
[62] Lunyak, Vlctoria & Kennedy, Brian. (2011). Aged Worms Erase Epigenetic History. Cell metabolism. 14. 147-8. 10.1016/j.cmet.2011.07.008.
[63] Pardee, Arthur. (2015). Bioregulation. Journal of Cellular Physiology. 230. 10.1002/jcp.25059.
[64] Schuetze, Katherine & Koch, Keith & McKinsey, Timothy. (2016). The potential of targeting epigenetic regulators for the treatment of fibrotic cardiac diseases. Future Medicinal Chemistry. 8. 10.4155/fmc-2016-0144.
[65] Eme, Laura & Spang, Anja & Lombard, Jonathan & Stairs, Courtney & Ettema, Thijs. (2017). Archaea and the origin of eukaryotes. Nature Reviews Microbiology. 16. 10.1038/nrmicro.2017.154.
[66] Brown, Michael. (2012). Curvature Forces in Membrane Lipid-Protein Interactions. Biochemistry. 51. 10.1021/bi301332v.
[67] Schwartz, Jeffrey. (2021). Evolution, systematics, and the unnatural history of mitochondrial DNA. Mitochondrial DNA Part A. 32. 1-26. 10.1080/24701394.2021.1899165.
[68] Garg, Abhishek & Maes, Hannelore & van Vliet, Alexander & Agostinis, Patrizia. (2015). Targeting the hallmarks of cancer with therapy-induced endoplasmic reticulum (ER) stress. Molecular & Cellular Oncology. 2. e975089. 10.4161/23723556.2014.975089.
[69] Caliari, Adriano & Xu, Jian & Yomo, Tetsuya. (2021). The requirement of cellularity for abiogenesis. Computational and Structural Biotechnology Journal. 19. 10.1016/j.csbj.2021.04.030.
[70] Sato, Naoki. (2020). Mereschkowsky, Founder of Endosymbiotic Hypothesis. 10.1007/978-981-15-1161-5_2
[71] McKinnon, Lucas & Theg, Steven. (2019). Information in the N-terminal region of targeting peptides determines the specificity of protein targeting to chloroplasts or mitochondria. Molecular Plant. 12. 10.1016/j.molp.2019.05.004.
[72] Veloz, Tomas & Flores, Daniela. (2021). Toward endosymbiosis modeling using reaction networks. Soft Computing. 25. 10.1007/s00500-020-05530-2.
[73] Peters, Winfried & Jensen, Kaare & Stone, Howard & Knoblauch, Michael. (2020). Plasmodesmata and the problems with size: Interpreting the confusion. Journal of Plant Physiology. 257. 153341. 10.1016/j.jplph.2020.153341.
[74] Shepherd, Jack & Payne-Dwyer, Alexander & Lee, Ji-Eun & Syeda, Aisha & Leake, Mark. (2021). Combining single-molecule super-resolved localization microscopy with fluorescence polarization imaging to study cellular processes. Journal of Physics: Photonics. 3. 10.1088/2515-7647/ac015d.
[75] Zeng, Zhiping & Ma, Jing & Canhua, Xu. (2020). Correlative cross-cumulant and radiality microscopy for multicolor super-resolution subcellular imaging. Photonics Research. 8. 10.1364/PRJ.387582
[76] Watson, Zoe & Ward, Fred & Méheust, Raphaël & Ad, Omer & Schepartz, Alanna & Banfield, Jillian & Cate, Jamie. (2020). Structure of the bacterial ribosome at 2 Å resolution. 10.7554/eLife.60482.sa2.
[77] Panagaki, Dimitra & Croft, Jacob & Keuenhof, Katharina & Larsson Berglund, Lisa & Andersson, Stefanie & Kohler, Verena & Büttner, Sabrina & Tamás, Markus & Nystrom, Thomas & Neutze, Richard & Höög, Johanna. (2021). Nuclear envelope budding is a response to cellular stress. Proceedings of the National Academy of Sciences. 118. e2020997118. 10.1073/pnas.2020997118.
[78] Lehmann, Kathrin & Shayegan, Marjan & Blab, Gerhard & Forde, Nancy. (2020). Optical Tweezers Approaches for Probing Multiscale Protein Mechanics and Assembly. Frontiers in Molecular Biosciences. 7. 577314. 10.3389/fmolb.2020.577314.
[79] Curcic, Sanja & Tiapko, Oleksandra & Groschner, Klaus. (2019). Photopharmacology and opto-chemogenetics of TRPC channels-some therapeutic visions. Pharmacology & Therapeutics. 200. 10.1016/j.pharmthera.2019.04.003.
[80] Hansel, Catherine & Holme, Margaret & Gopal, Sahana & Stevens, Molly. (2019). Advances in high-resolution microscopy for the study of intracellular interactions with biomaterials. Biomaterials. 226. 119406. 10.1016/j.biomaterials.2019.119406.
[81] Li, Xia & Park, Donghyun & Chang, Yunjie & Radhakrishnan, Abhijith & Wu, Hangjun & Wang, Pei & Liu, Jun. (2020). A mammalian system for high-resolution imaging of intact cells by cryo-electron tomography. Progress in Biophysics and Molecular Biology. 160. 10.1016/j.pbiomolbio.2020.09.005.
[82] Santos de Macedo, Cristiana & Anderson, David & Schey, Kevin. (2017). MALDI (matrix assisted laser desorption ionization) Imaging Mass Spectrometry (IMS) of skin: Aspects of sample preparation. Talanta. 174. 10.1016/j.talanta.2017.06.018.
[83] Mattiazzi, Mojca & Yeung, Clarence & Friesen, Helena & Boone, Charles & Andrews, Brenda. (2021). Single-cell image analysis to explore cell-to-cell heterogeneity in isogenic populations. Cell Systems. 12. 608-621. 10.1016/j.cels.2021.05.010.
[84] Liu, Yaoping & Xu, Han & LI, TINGYU & Wang, Wei. (2021). Microtechnology enabled filtration-based liquid biopsy: challenges and practical considerations. Lab on a Chip. 21. 10.1039/D0LC01101K.
[85] Qu, Yufu & Hao, Jialin & Peng, Renju. (2019). Machine-learning models for analyzing TSOM images of nanostructures. Optics Express. 27. 33978. 10.1364/OE.27.033978.
[86] Michalska, Karolina & Joachimiak, Andrzej. (2021). Structural Genomics and the Protein Data Bank. Journal of Biological Chemistry. 296. 100747. 10.1016/j.jbc.2021.100747.
[87] Hartwell, Leland & Hopfield, John & Leibler, Stanislas & Murray, Andrew. (2000). From Molecular to Modular Cell Biology. Nature. 402. C47-52. 10.1038/35011540.
[88] Promislow, Daniel & McCormick, Mark. (2017). Recent Advances in the Systems Biology of Aging. Antioxidants & Redox Signaling. 29. 10.1089/ars.2017.7367.
[89] Lazebnik, Yuri. (2005). Can a biologist fix a radio? -- Or, what I learned while studying apoptosis, (Cancer Cell. 2002 Sep;2(3):179-82). Biochemistry. Biokhimii?a. 69. 1403-6. 10.1007/s10541-005-0013-7.
[90] Csete, Marie & Doyle, John. (2002). Reverse Engineering of Biological Complexity. Science (New York, N.Y.). 295. 1664-9. 10.1126/science.1069981.
[91] Gerardin, Jaline & Reddy, Nishith & Lim, Wendell. (2019). The Design Principles of Biochemical Timers: Circuits that Discriminate between Transient and Sustained Stimulation. Cell Systems. 9. 10.1016/j.cels.2019.07.008.
[92] Mast, Fred & Rachubinski, Richard & Aitchison, John. (2020). Peroxisome prognostications: Exploring the birth, life, and death of an organelle. The Journal of Cell Biology. 219. e201912100. 10.1083/jcb.201912100.
[93] Keating, Sarah & Waltemath, Dagmar & König, Matthias & Zhang, Fengkai & Dräger, Andreas & Chaouiya, Claudine & Bergmann, Frank & Finney, Andrew & Gillespie, Colin & Helikar, Tomas & Hoops, Stefan & Malik Sheriff, Rahuman & Moodie, Stuart & Moraru, Ion & Myers, Chris & Naldi, Aurélien & Olivier, Brett & Sahle, Sven & Schaff, James. (2020). SBML Level 3: an extensible format for the exchange and reuse of biological models. Molecular Systems Biology. 16. 10.15252/msb.20199110.
[94] Dani, Diksha & Sainis, Jayashree. (2007). Modularity: A new perspective in biology. Indian journal of biochemistry & biophysics. 44. 133-9.
[95] Yaguchi, Shunsuke & Yaguchi, Junko & Suzuki, Haruka & Kinjo, Sonoko & Kiyomoto, Masato & Ikeo, Kazuho & Yamamoto, Akane. (2020). Current Biology. Current Biology.
[96] Torras, Núria & Garcia-Diaz, Maria & Fernández-Majada, Vanesa & Martinez, Elena. (2018). Mimicking Epithelial Tissues in Three-Dimensional Cell Culture Models. Frontiers in Bioengineering and Biotechnology. 6. 197. 10.3389/fbioe.2018.00197.
[97] Levin, Michael. (2021). Bioelectric signaling: Reprogrammable circuits underlying embryogenesis, regeneration, and cancer. Cell. 184. 10.1016/j.cell.2021.02.034.
[98] Gilbert, Scott & Corfe, Ian. (2013). Turtle Origins: Picking Up Speed. Developmental cell. 25. 326-328. 10.1016/j.devcel.2013.05.011
[99] Kammerer, Theresa. (2021). Visualization of the Extracellular Matrix (ECM) by fluorescent tagging of ECM components in mouse.
[100] Rozario, Tania & Newmark, Phillip. (2018). Prospecting for Planarian Pluripotency. Cell. 173. 1566-1567. 10.1016/j.cell.2018.05.062.
[101] Friedman, Adam & Tucker, George & Singh, Rohit & Yan, Dong & Arunachalam, Vinayagam & Hu, Claire(Yanhui) & Binari, Richard & Hong, Pengyu & Sun, Xiaoyun & Porto, Maura & Pacifico, Svetlana & Murali, Thilakam & Finley, Russell & Asara, John & Berger, Bonnie & Perrimon, Norbert. (2011). Proteomic and Functional Genomic Landscape of Receptor Tyrosine Kinase and Ras to Extracellular Signal-Regulated Kinase Signaling. Science signaling. 4. rs10. 10.1126/scisignal.2002029.
[102] Yeh, Chiuan-Ren & Bingham, Grace & Shetty, Jagathpala & Hu, Ping & Barker, Thomas. (2021). Decellularized Extracellular Matrix (ECM) as a Model to Study Fibrotic ECM Mechanobiology. 10.1007/978-1-0716-1382-5_18.
[103] Chighizola, Matteo & Dini, Tania & Lenardi, Cristina & Milani, Paolo & Podestà, Alessandro & Schulte, Carsten. (2019). Mechanotransduction in Neuronal Cell Development and Functioning. 10.20944/preprints201909.0241.v1.
[104] Conrad, Christina & Moore, Kaitlin & Polacheck, William & Rizvi, Imran & Scarcelli, Giuliano. (2021). Mechanical Modulation of Tumor Nodules under Flow. IEEE Transactions on Biomedical Engineering. PP. 1-1. 10.1109/TBME.2021.3092641.
[105] Barbosa, Yago & Melo, Wanderson & Rodrigues, Huanna & Neto, Napoleão. (2020). Aplicabilidade da estereologia e microscopia de força atômica à pesquisa dermatológica veterinária (revisão de literatura). Jornal Interdisciplinar de Biociências. 4. 16. 10.26694/jibi.v4i1.8596.
[106] Pasakarnis, Laurynas. (2016). Analysis of acto-myosin network remodelling: towards a comprehensive understanding of tissue closure event
[107] McIntosh, Richard & Hays, Thomas. (2016). A Brief History of Research on Mitotic Mechanisms. Biology. 5. 10.3390/biology5040055
[108] Lang, Xiaoqi & Welsher, Kevin. (2020). Mapping solvation heterogeneity in live cells by hyperspectral stimulated Raman scattering microscopy. The Journal of Chemical Physics. 152. 174201. 10.1063/1.5141422.
[109] Dolnik, Olga & Gerresheim, Gesche & Biedenkopf, Nadine. (2021). New Perspectives on the Biogenesis of Viral Inclusion Bodies in Negative-Sense RNA Virus Infections. Cells. 10. 1460. 10.3390/cells10061460
[110] Enukashvily, Natella & Dobrynin, Mikhail & Chubar, Anna. (2021). RNA-seeded membraneless bodies: Role of tandemly repeated RNA. Advances in Protein Chemistry and Structural Biology. 10.1016/bs.apcsb.2020.12.007.
[111] Stroberg, Wylie & Schnell, Santiago. (2020). Concentration Sensing in Crowded Environments. 10.1101/2020.10.02.324129.
[112] Lang, Xiaoqi & Welsher, Kevin. (2020). Mapping solvation heterogeneity in live cells by hyperspectral stimulated Raman scattering microscopy. The Journal of Chemical Physics. 152. 174201. 10.1063/1.5141422.
[113] Altenburg, Wiggert & Yewdall, N. Amy & Vervoort, Daan & Stevendaal, Marleen & Mason, Alexander & Hest, Jan. (2020). Programmed spatial organization of biomacromolecules into discrete, coacervate-based protocells. Nature Communications. 11. 10.1038/s41467-020-20124-0.
[114] Breindel, Leonard & Yu, Jianchao & Burz, David & Shekhtman, Alexander. (2020). Intact ribosomes drive the formation of protein quinary structure. PLOS ONE. 15. e0232015. 10.1371/journal.pone.0232015.
[115] Bagatolli, Luis & Stock, Roberto. (2021). Lipids, membranes, colloids and cells: A long view. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1863. 183684. 10.1016/j.bbamem.2021.183684.
[116] Assaiya, Anshul & Burada, Ananth & Dhingra, Surbhi & Kumar, Janesh. (2021). An overview of the recent advances in cryo-electron microscopy for life sciences. Emerging Topics in Life Sciences. 5. 10.1042/ETLS20200295.
[117] Ristovski, Miliça & Farhat, Danny & Bancud, Shelly & Lee, Jyh-Yeuan. (2021). Lipid Transporters Beam Signals from Cell Membranes. Membranes. 11. 562. 10.3390/membranes11080562.
[118] Wu, Erzhong & Guo, Xuzhen & Teng, Xueyi & Zhang, Ruijin & Li, Fahui & Cui, Ya & Zhang, Dongdong & Liu, Qinghua & Luo, Jianjun & Wang, Jiangyun & Chen, Runsheng. (2021). Discovery of Plasma Membrane-Associated RNAs through APEX-seq. Cell Biochemistry and Biophysics. 10.1007/s12013-021-00991-0.
[119] Costa, Ana & Sousa, Monica. (2021). The role of the membrane-associated periodic skeleton in axons. Cellular and Molecular Life Sciences. 78. 10.1007/s00018-021-03867-x.
[120] Renart, María & Giudici, Ana & Díaz-García, Clara & Molina Gallego, María & Morales, Andrés & González-Ros, José & Poveda, José. (2020). Modulation of Function, Structure and Clustering of K+ Channels by Lipids: Lessons Learnt from KcsA. International Journal of Molecular Sciences. 21. 2554. 10.3390/ijms21072554.
[121] Schmid, Friederike & Dolezel, Stefan & Lenz, Olaf & Meinhardt, Sebastian. (2014). On ripples and rafts: Curvature induced nanoscale structures in lipid membranes. Journal of Physics: Conference Series. 487. 10.1088/1742-6596/487/1/012004.
[122] Friedrich, Dhana. (2020). Oscillatory transcription factors and stochastic gene expression. 10.18452/22053.
[123] Pejchar, P?emysl & Sekereš, Juraj & Novotny, Ond?ej & Zárský, Viktor & Potocký, Martin. (2020). Functional analysis of phospholipase Dδ family in tobacco pollen tubes. The Plant Journal. 103. 10.1111/tpj.14720.
[124] Santos, Herbert & Nozaki, Tomoyoshi. (2021). Interorganellar communication and membrane contact sites in protozoan parasites. Parasitology International. 83. 102372. 10.1016/j.parint.2021.102372.
[125] Xie, Beichen & Panagiotou, Styliani & Cen, Jing & Gilon, Patrick & Bergsten, Peter & Idevall-Hagren, Olof. (2021). The endoplasmic reticulum-plasma membrane tethering protein TMEM24 is a regulator of cellular Ca2+ homeostasis. 10.1101/2021.06.23.449694.
[126] uang, Tianzao & Chen, Yingxian & Zeng, Yile & Xu, Chaoyang & Huang, Jinzhong & Hu, Weipeng & Chen, Xiangrong & Fu, Huangde. (2021). Long non-coding RNA PSMA3-AS1 promotes glioma progression through modulating the miR-411-3p/HOXA10 pathway. BMC Cancer. 21. 10.1186/s12885-021-08465-5.
[127] Mazzarello, Paolo. (1999). A unifying concept: the history of cell theory. Nature cell biology. 1. E13-5. 10.1038/8964.
[128] Chialvo, Dante. (2002). Physiology - Unhealthy surprises. Nature. 419. 263. 10.1038/419263a
[129] Goldbeter, Albert. (2002). Computational approaches to cellular rhythms. Nature. 420. 238-45. 10.1038/nature01259
[130] Wimsatt, William. (2002). Using False Models to Elaborate Constraints on Processes: Blending Inheritance in Organic and Cultural Evolution. Philosophy of Science. 69. 10.1086/341764.
[131] Hasty, Jeff & Mcmillen, David & Collins, J. (2002). Engineered Gene Circuits. Nature. 420. 224-30. 10.1038/nature01257.
[132] Karlebach, Guy & Shamir, Ron. (2008). Modelling and analysis of gene regulatory networks. Nature reviews. Molecular cell biology. 9. 770-80. 10.1038/nrm2503.
[133] Lewis, John & Glass, Leon. (1992). Nonlinear Dynamics and Symbolic Dynamics of Neural Networks. Neural Computation. 4. 621-642. 10.1162/neco.1992.4.5.621.
[134] Guarente, Leonard & Kenyon, Cynthia. (2000). Guarente, L. & Kenyon, C. Genetic pathways that regulate ageing in model organisms. Nature 408, 255-262. Nature. 408. 255-62. 10.1038/35041700
[135] Seroude, Laurent. (2002). Differential Gene Expression and Aging. TheScientificWorldJournal. 2. 618-31. 10.1100/tsw.2002.135.
[136] Rao, Potu & Johnson, Tangmit. (1970). Mammalian Cell Fusion : Studies on the Regulation of DNA Synthesis and Mitosis. Nature. 225. 159-64. 10.1038/225159a0.
[137] Masui, Yoshio. (2001). The elusive cytostatic factor in the animal egg. Nature reviews. Molecular cell biology. 1. 228-32. 10.1038/35043096.
[138] Reimann, Julie & Freed, Ellen & Hsu, Jerry & Kramer, Edgar & Peters, Jan-Michael & Jackson, Peter. (2001). Emi1 Is a Mitotic Regulator that Interacts with Cdc20 and Inhibits the Anaphase Promoting Complex. Cell. 105. 645-55. 10.1016/S0092-8674(01)00361-0.
[139] Pardee, Arthur. (1974). A Restriction Point for Control of Normal Animal Cell Proliferation. Proceedings of the National Academy of Sciences of the United States of America. 71. 1286-90. 10.1073/pnas.71.4.1286.
[140] Nurse, Paul. (1975). Genetic control of cell size at cell division in yeast. Nature. 256. 547-51. 10.1038/256547a0.
[141] Clarke, Louise & Carbon, John. (1980). Isolation of a yeast centromere and construction of functional small circular chromosomes. Nature. 287. 504-9. 10.1038/287504a0.
[142] Evans, Tom & Rosenthal, Eric & Youngblom, Jim & Distel, Daniel & Hunt, Tim. (1983). Cyclin: A protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell. 33. 389-96. 10.1016/0092-8674(83)90420-8.
[143] Murray, Andrew & Kirschner, Marc. (1989). Cyclin synthesis drives the early embryonic cell cycle. Nature. 339. 10.1038/339275a0.
[144] Fagrelius, Thomas & Livingston, Dennis. (1984). Location of DNAse I-sensitive cleavage sites in the Yeast 2µ plasmid DNA chromosome. Journal of molecular biology. 173. 1-13. 10.1016/0022-2836(84)90400-5.
[145] Brümmer, Anneke & Salazar, Carlos & Zinzalla, Vittoria & Alberghina, Lilia & Höfer, Thomas. (2010). Mathematical Modelling of DNA Replication Reveals a Trade-off between Coherence of Origin Activation and Robustness against Rereplication. PLoS computational biology. 6. e1000783. 10.1371/journal.pcbi.1000783.
[146] Weinert, A. & T. Hartwell , L. H. RAD9 gen controla la respuesta del ciclo celular al daño del ADN en Saccharomyces cerevisiae. Ciencia 241, 317-322.
[147] Villagómez, Mario & Vélez Paez, Jorge Luis & González, Fernando & Velarde, Gustavo & Páez, Pablo & Aguayo Moscoso, Santiago. (2020). El ciclo celular, sus mecanismos de regulación y reparación, indispensables para el mantenimiento de la vida. The cell cycle and its regulation and repair mechanisms essential for the maintenance of life.. 4. 1-25.
[148] Lieberman, Howard B & Hopkins, Kevin M & Nass, Monica & Demetrick, Douglas & Davey, Scott. (1996). A human homolog of the Schizosaccharomyces pombe rad9+ checkpoint control gene. Proceedings of the National Academy of Sciences of the United States of America. 93. 13890-5. 10.1073/pnas.93.24.13890.
[149] Whyte, P.F.M. & Buchkovich, Karen & Horowitz, Jonathan & Friend, Stephen & Raybuck, Margaret & Weinberg, Robert & Harlow, Ed. (1988). Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature. 334. 124-9. 10.1038/334124a0.
[150] Baker, S & Fearon, E & Nigro, Janice & Hamilton, S & Preisinger, AC & Jessup, John & Vantuinen, Peter & Ledbetter, David & Barker, David & Nakamura, Yasuhiro & White, Rainelle & Vogelstein, Bert. (1989). Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science (New York, N.Y.). 244. 217-21. 10.1126/science.2649981.
[151] Loewer, Alexander & Batchelor, Eric & Gaglia, Giorgio & Lahav, Galit. (2010). Basal Dynamics of p53 Reveal Transcriptionally Attenuated Pulses in Cycling Cells. Cell. 142. 89-100. 10.1016/j.cell.2010.05.031.
[152] Edgar, Bruce & O'Farrell, Patrick. (1989). Genetic Control of Cell Division Patterns in the Drosophila Embryo. Cell. 57. 177-87. 10.1016/0092-8674(89)90183-9.
[153] Hoyt, M.Andrew & Totis, Laura & Roberts, B.Tibor. (1991). S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 66. 507-17. 10.1016/0962-8924(91)90104-H.
Li, Rong & Murray, Andrew. (1991). Feedback control of mitosis in budding yeast. Cell. 66. 519-31. 10.1016/0092-8674(81)90015-5.
[154] Matsushime, Hitoshl & Roussel, Martine & Ashmun, Richard & Sherr, Charles. (1991). Matsushime, H, Roussel, MF, Ashmun, RA & Sherr, CJColony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell 65: 701-713. Cell. 65. 701-13. 10.1016/0092-8674(91)90101-4.
Xiong, Yue & Connolly, Tim & Futcher, Bruce & Beach, David. (1991). Human D-Type cyclin. Cell. 65. 691-9. 10.1016/0092-8674(91)90100-D.
[155] Koff, Andrew & Cross, Fred & Fisher, Alfred & Schumacher, Jill & Leguellec, Katherine & Philippe, Michel & Roberts, James. (1991). Human Cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell. 66. 1217-28. 10.1016/0092-8674(91)90044-Y.
[156] Greene DM, Hsu DW, Peras CJ.Control de los niveles de ciclina C durante el desarrollo de Dictyostelium. PLoS ONE. (5): e10543. (2010)
[157] Motokura, Toru & Bloom, Theodora & Kim, Hyung & Jüppner, Harald & Ruderman, Joan & Kronenberg, Henry & Arnold, Andrew. (1991). A novel cyclin encoded BCL1-linked candidate oncogene. Nature. 350. 512-5. 10.1038/350512a0.
[158] Glotzer, Michael & Murray, Andrew & Kirschner, Marc. (1991). Cyclin Is Degraded by the Ubiquitin Pathway. Nature. 349. 132-8. 10.1038/349132a0.
[159] Richly, Holger & Rape, Michael & Braun, Sigurd & Rumpf, Sebastian & Hoege, Carsten & Jentsch, Stefan. (2005). A Series of Ubiquitin Binding Factors Connects CDC48/p97 to Substrate Multiubiquitylation and Proteasomal Targeting. Cell. 120. 73-84. 10.1016/j.cell.2004.11.013.
Seoane, Joan & Le, Hong-Van & Shen, Lijian & Anderson, Stewart & Massagué, Joan. (2004). Integration of Smad and Forkhead Pathways in the Control of Neuroepithelial and Glioblastoma Cell Proliferation. Cell. 117. 211-23. 10.1016/S0092-8674(04)00298-3.
[160] Schwob, Etienne & Böhm, Thomas & Mendenhall, Michael & Nasmyth, Kim. (1994). The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 79. 233-44. 10.1016/0092-8674(94)90193-7.
Li, Wei & You, Liru & Cooper, Jonathan & Schiavon, Gaia & Pepe-Caprio, Angela & Zhou, Lu & Ishii, Ryohei & Giovannini, Marco & Hanemann, Clemens & Long, Stephen & Erdjument-Bromage, Hediye & Zhou, Pengbo & Tempst, Paul & Giancotti, Filippo. (2010). Merlin/NF2 Suppresses Tumorigenesis by Inhibiting the E3 Ubiquitin Ligase CRL4DCAF1 in the Nucleus. Cell. 140. 477-90. 10.1016/j.cell.2010.01.029.
[161] Uhlmann, Frank. (2014). A silent revolution in chromosome biology. Nature reviews. Molecular cell biology. 15. 10.1038/nrm3817.
[162] Separase, Polo Kinase, the Kinetochore Protein Slk19, and Spo12 Function in a Network that Controls Cdc14 Localization during Early Anaphase. Cell. 108. 207-20. 10.1016/S0092-8674(02)00618-9.
[163] Ishiyama, Noboru & Lee, Seung-Hye & Liu, Shuang & Li, Guang-Yao & Smith, Matthew & Reichardt, Louis & Ikura, Mitsu. (2010). Dynamic and Static Interactions between p120 Catenin and E-Cadherin Regulate the Stability of Cell-Cell Adhesion. Cell. 141. 117-28. 10.1016/j.cell.2010.01.017.
[164] Drost, Jarno & Agami, Reuven. (2009). Transformation Locked in a Loop. Cell. 139. 654-6. 10.1016/j.cell.2009.10.035.
[165] Ma, Yan & Fiering, Steven & Black, Candice & Liu, Xi & Yuan, Ziqiang & Memoli, Vincent & Robbins, David & Bentley, Heather & Tsongalis, Gregory & Demidenko, Eugene & Freemantle, Sarah & Dmitrovsky, Ethan. (2007). Transgenic cyclin E triggers dysplasia and multiple pulmonary adenocarcinomas. Proceedings of the National Academy of Sciences of the United States of America. 104. 4089-94. 10.1073/pnas.0606537104.
[166] Doucet, Christine & Talamas, Jessica & Hetzer, Martin. (2010). Cell Cycle-Dependent Differences in Nuclear Pore Complex Assembly in Metazoa. Cell. 141. 1030-41. 10.1016/j.cell.2010.04.036.
[167] Sherr, Charles & Roberts, James. (1999). CDK inhibitor: Positive and negative regulators of G1-phase progression. Genes & development. 13. 1501-12. 10.1101/gad.13.12.1501.
[168] Cooper, Stephen. (2000). The Continuum model and G1-control of the mammalian cell cycle. Progress in cell cycle research. 4. 27-39. 10.1007/978-1-4615-4253-7_3.
[169] Malumbres, Marcos & Barbacid, Mariano. (2002). To cycle or not to cycle: A critical decision in cancer. Nature reviews. Cancer. 1. 222-31. 10.1038/35106065.
[170] Shaffer, David & Viale, Agnes & Ishiwata, Ryota & Leversha, Margaret & Olgac, Semra & Manova, Katia & Satagopan, Jaya & Scher, Howard & Koff, Andrew. (2005). Evidence for a p27 tumor suppressive function independent of its role regulating cell proliferation in the prostate. Proceedings of the National Academy of Sciences of the United States of America. 102. 210-5. 10.1073/pnas.0407362102.
[171] Watanabe, Nobumoto & Arai, Harumi & Iwasaki, Jun-Ichi & Shiina, Masaaki & Ogata, Kazuhiro & Hunter, Tony & Osada, Hiroyuki. (2005). Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proceedings of the National Academy of Sciences of the United States of America. 102. 11663-8. 10.1073/pnas.0500410102.
[172] Tsutsui, Tateki & Hesabi, Bahar & Moons, David & Pandolfi, Pier & Hansel, Kimberly & Koff, Andrew & Kiyokawa, Hiroaki. (1999). Targeted Disruption of CDK4 Delays Cell Cycle Entry with Enhanced p27Kip1 Activity. Molecular and cellular biology. 19. 7011-9. 10.1128/MCB.19.10.7011.
[173] Cheng, Mangeng & Sexl, Veronika & Sherr, Charles & Roussel, Martine. (1998). Cheng M, Sexl V, Sherr CJ, Roussel MF.. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc Natl Acad Sci USA 95: 1091-1096. Proceedings of the National Academy of Sciences of the United States of America. 95. 1091-6. 10.1073/pnas.95.3.1091.
[174] Lee, Hyeong-Min. (2010). The Essential Roles Of CKIδ/ε In The Mammalian Circadian Clock.
[175] Okamoto, Kengo & Sagata, Noriyuki. (2007). Mechanism for inactivation of the mitotic inhibitory kinase Wee1 at M phase. Proceedings of the National Academy of Sciences of the United States of America. 104. 3753-8. 10.1073/pnas.0607357104.
Passer, Brent & Nancy-Portebois, Vanessa & Amzallag, Nathalie & Prieur, Sylvie & Cans, Christophe & Climens, Aude & Fiucci, Giusy & Bouvard, Véronique & Tuynder, Marcel & Susini, Laurent & Morchoisne, Stéphanie & Crible, Virginie & Lespagnol, Alexandra & Dausset, Jean & Oren, Moshe & Amson, Robert & Telerman, Adam. (2003). The p53-inducible TSAP6 gene product regulates apoptosis and the cell cycle and interacts with Nix and the Myt1 kinase. Proceedings of the National Academy of Sciences of the United States of America. 100. 2284-9. 10.1073/pnas.0530298100.
[176] Sotillo, Rocio & García, Juan & Ortega, Sagrario & Martín, Javier & Dubus, Pierre & Barbacid, Mariano & Malumbres, Marcos. (2001). Invasive melanoma in Cdk4-targeted mice. Proceedings of the National Academy of Sciences of the United States of America. 98. 13312-7. 10.1073/pnas.241338598.
[177] Moustakas, Aristidis & Kardassis, Dimitris. (1998). Regulation of the Human p21/WAF1/Cip1 Promoter in Hepatic Cells by Functional Interactions between Sp1 and Smad Family Members. Proceedings of the National Academy of Sciences of the United States of America. 95. 6733-8. 10.1073/pnas.95.12.6733.
[178] Marches, Radu & Hsueh, Robert & Uhr, Jonathan. (1999). Cancer dormancy and cell signaling: Induction of p21waf1 initiated by membrane IgM engagement increases survival of B lymphoma cells. Proceedings of the National Academy of Sciences. 96. 10.1073/pnas.96.15.8711.
[179] Lee, Y.J. & Hahn, Y. & Park, J. & Park, C. & Hyun, B. & Chung, J.H.. (1999). Mus musculus molossinus retinoblastoma-like 1 (Rbl1) mRNA, partial sequence.
[180] Morgan, David. (1997). Morgan DO. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu Rev Cell Dev Biol13: 261-291. Annual review of cell and developmental biology. 13. 261-91. 10.1146/annurev.cellbio.13.1.261.
Adams, Peter. (2001). Adams, P. D. Regulation of the retinoblastoma tumor suppressor protein by cyclin/CDKs. Biochim. Biophys. Acta 1471, 123-133. Biochimica et biophysica acta. 1471. M123-33. 10.1016/S0304-419X(01)00019-1.
[181] Farkas, Thomas & Hansen, Klaus & Holm, Karin & Lukas, Jiri & Bartek, Jiri. (2002). Distinct Phosphorylation Events Regulate p130- and p107-mediated Repression of E2F-4. The Journal of biological chemistry. 277. 26741-52. 10.1074/jbc.M200381200.
[182] Harbour, J William & Dean, D. (2000). Harbour JW & Dean DC. The Rb/E2F pathway: Expanding roles and emerging paradigms. Genes Dev14: 2393-2409. Genes & development. 14. 2393-409. 10.1101/gad.813200.
Koga, Minori & Ishiguro, Hiroki & Yazaki, Saori & Horiuchi, Yasue & Arai, Makoto & Niizato, Kazuhiro & Iritani, Shuji & Itokawa, Masanari & Inada, Toshiya & Iwata, Nakao & Ozaki, Norio & Ujike, Hiroshi & Kunugi, Hiroshi & Sasaki, Tsukasa & Takahashi, Makoto & Watanabe, Yuichiro & Someya, Toshiyuki & Kakita, Akiyoshi & Takahashi, Hitoshi & Arinami, Tadao. (2009). Involvement of SMARCA2/BRM in the SWI/SNF chromatin-remodeling complex in schizophrenia. Human molecular genetics. 18. 2483-94. 10.1093/hmg/ddp166.
[183] Nevins, Joseph. (2001). The Rb/E2F pathway and cancer. Human molecular genetics. 10. 699-703. 10.1093/hmg/10.7.699.
[184] Negri, Gloria & Rodríguez, Cristina. (2017). EP300 (E1A binding protein p300). Atlas of Genetics and Cytogenetics in Oncology and Haematology. 10.4267/2042/62485.
[185] Chan, Homan & Krstic-Demonacos, Marija & Smith, Linda & Demonacos, Costas & Thangue, Nicholas. (2001). Acetylation control of the retinoblastoma tumor-suppressor. Nature cell biology. 3. 667-74. 10.1038/35083062.
[186] Malumbres, Marcos & Pellicer, Angel. (1998). RAS pathways to cell cycle control and cell transformation. Frontiers in bioscience : a journal and virtual library. 3. d887-912. 10.2741/A331.
[187] Sicinski, Piotr & Donaher, Joana & Parker, Susan & Li, Tiansen & Fazeli, Amin & Gardner, Humphrey & Haslam, Sandra & Bronson, Roderick & Elledge, Stephen & Weinberg, Robert. (1995). Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, Weinberg RACyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 82: 621-630. Cell. 82. 621-30. 10.1016/0092-8674(95)90034-9.
[188] Malliri, Angeliki & Kammen, Rob & Clark, Kristopher & Valk, Maarten & Michiels, Frits & Collard, John. (2002). Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature. 417. 867-71. 10.1038/nature00848.
[189] Hosokawa, Yoshitaka & Arnold, Andrew. (1996). Cyclin D1/PRAD1 as a central target in oncogenesis. The Journal of laboratory and clinical medicine. 127. 246-52. 10.1016/S0022-2143(96)90092-X.
[190] Sheer, C.J.. (2000). The Pezcoller lecture: Cancer cell cycles revisited. Cancer Res.. 60. 3689-3695.
[191] Roovers, Kristin & Assoian, Richard. (2000). Integrating the MAP kinase signal into the G1 phase cell cycle machinery. BioEssays : news and reviews in molecular, cellular and developmental biology. 22. 818-26. 10.1002/1521-1878(200009)22:9<818::AID-BIES7>3.0.CO;2-6.
[192] Leonard, G. & Cynthia, K.. (2010). Genetic pathways that regulate ageing in model organisms. Nature.. 408. 255-262.
[193] Mikkola, Hanna & Klintman, Jenny & Yang, Haidi & Hock, Hanno & Schlaeger, Thorsten & Fujiwara, Yuko & Orkin, Stuart. (2003). Haematopoietic stem cells retain long-term repopulating activity and multipotency in the absence of stem-cell leukaemia SCL/tal-1 gene. Nature. 421. 547-51. 10.1038/nature01345.
[194] Bakkenist, Christopher & Kastan, Michael. (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 421. 499-506. 10.1038/nature01368.
[195] Duman-Scheel, Molly & Weng, Li & Xin, Shijie & Du, Wei. (2002). Duman-Scheel M, Weng L, Xin S, Du W.. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature 417: 299-304. Nature. 417. 299-304. 10.1038/417299a.
[196] Gomez-Roman, Natividad & Grandori, Carla & Eisenman, Robert & White, Robert. (2003). Direct activation of RNA polymerase III transcription by c-MYC. Nature. 421. 290-4. 10.1038/nature01327.
[197] Johnston, Jennifer & Dalton, Muffie & Gurney, Mark & Kopito, Ron. (2000). Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 97. 12571-6. 10.1073/pnas.220417997.
[198] Rondon, Michelle & Raffel, Sandra & Goodman, Robert & Handelsman, Jo. (1999). Toward functional genomics in bacteria: Analysis of gene expression in Escherichia coli from a bacterial artificial chromosome library of Bacillus cereus. Proceedings of the National Academy of Sciences of the United States of America. 96. 6451-5. 10.1073/pnas.96.11.6451.
[199] Murphy, Paul & Bolduc, Jill & Gallaher, Jasmine & Stoddard, Barry & Baker, David. (2009). Alteration of enzyme specificity by computational loop remodeling and design. Proceedings of the National Academy of Sciences of the United States of America. 106. 9215-20. 10.1073/pnas.0811070106.
[200] Au-Yeung, Ho Yu & Panto?, Dan & Sanders, Jeremy. (2009). Molecular recognition and self-assembly special feature: Dynamic combinatorial synthesis of a catenane based on donor-acceptor interactions in water. Proceedings of the National Academy of Sciences of the United States of America. 106. 10466-70. 10.1073/pnas.0809934106.
[201] Trimarchi, Jeffrey & Fairchild, Brian & RL, Verona & Moberg, Ken & Andon, Nancy & Lees, Jacqueline. (1998). E2F-6, a Member of the E2F Family that can Behave as a Transcriptional Repressor. Proceedings of the National Academy of Sciences of the United States of America. 95. 2850-5. 10.1073/pnas.95.6.2850.
[202] Tazawa, Hiroshi & Tsuchiya, Naoto & Izumiya, Masash & Nakagama, Hitoshi. (2007). Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 104. 15472-7. 10.1073/pnas.0707351104.
[203] Sherr, Charles & Roberts, James. (1999). CDK inhibitor: Positive and negative regulators of G1-phase progression. Genes & development. 13. 1501-12. 10.1101/gad.13.12.1501.
[204] Dyson, N. (1998). Dyson N.. The regulation of E2F by pRB-family proteins. Genes Dev 12: 2245-2262. Genes & development. 12. 2245-62. 10.1101/gad.12.15.2245.
[205] Degregori, James & Leone, G & Ohtani, Kiyoshi & Miron, Alexander & Nevins, Joseph. (1996). E2F-1 accumulation bypasses a G1 arrest resulting from the inhibition of G1 cyclin-dependent kinase activity. Genes & development. 9. 2873-87. 10.1101/gad.9.23.2873.
[206] Giangrande, Paloma & Zhu, Wencheng & Schlisio, Susanne & Sun, Xin & Mori, Seiichi & Gaubatz, Stefan & Nevins, Joseph. (2005). A role for E2F6 in distinguishing G1/S- and G2/M-specific transcription. Genes & development. 18. 2941-51. 10.1101/gad.1239304.
[207] Russo, Giuseppe & Zamparelli, Alessandra & Howard-Claudio, Candace & Minimo, Corrado & Bellan, Cristiana & Carillo, Giovanna & Califano, Luigi & Leoncini, Lorenzo & Giordano, Antonio & Claudio, Pier Paolo. (2005). Expression of Cell Cycle-Regulated Proteins pRB2/p130, p107, E2F4, p27, and pCNA in Salivary Gland Tumors: Prognostic and Diagnostic Implications. Clinical cancer research : an official journal of the American Association for Cancer Research. 11. 3265-73. 10.1158/1078-0432.CCR-04-2508.
[208] Dou, Q & Markell, P & Pardee, A. (1992). Thymidine kinase transcription is regulated at G1/S by a complex that contains retinoblastoma-like protein and cdc2 kinase. Proceedings of the National Academy of Sciences of the United States of America. 89. 3256-60. 10.1073/pnas.89.8.3256.
[209] Girling, Rowena & Partridge, Janet & Bandara, Lasantha & Burden, Neil & Totty, Nicholas & Hsuan, J. & Thangue, Nicholas. (1993). A new component of the transcription factor DRTF1/E2F. Nature. 362. 83-7. 10.1038/362083a0.
[210] Berman, Seth & Yuan, Tina & Miller, Emily & Lee, Eunice & Caron, Alicia & Lees, Jacqueline. (2008). The Retinoblastoma Protein Tumor Suppressor Is Important for Appropriate Osteoblast Differentiation and Bone Development. Molecular cancer research : MCR. 6. 1440-51. 10.1158/1541-7786.MCR-08-0176.
[211] Helin, Kristian. (1999). Regulation of cell proliferation by the E2F transcription factors. Biochemical Society Transactions. 27. A64.1-A64. 10.1042/bst027a064.
[212] Trimarchi, Jeffrey & Fairchild, Brian & RL, Verona & Moberg, Ken & Andon, Nancy & Lees, Jacqueline. (1998). E2F-6, a Member of the E2F Family that can Behave as a Transcriptional Repressor. Proceedings of the National Academy of Sciences of the United States of America. 95. 2850-5. 10.1073/pnas.95.6.2850.
[213] Trimarchi, Jeffrey & Fairchild, Brian & RL, Verona & Moberg, Ken & Andon, Nancy & Lees, Jacqueline. (1998). E2F-6, a Member of the E2F Family that can Behave as a Transcriptional Repressor. Proceedings of the National Academy of Sciences of the United States of America. 95. 2850-5. 10.1073/pnas.95.6.2850.
[214] Krek, Wilhelm & Livingston, D.M. & Shirodkar, S. (1994). Binding to DNA and the Retinoblastoma Gene Product Promoted by Complex Formation of Different E2F Family Members. Science (New York, N.Y.). 262. 1557-60. 10.1126/science.8248803.
[215] Ivey-Hoyle, M & Conroy, R & Huber, H & Goodhart, P & Oliff, A & Heimbrook, D. (1994). Cloning and characterization of E2F-2, a novel protein with the biochemical properties of transcription factor E2F. Molecular and cellular biology. 13. 7802-12. 10.1128/MCB.13.12.7802.
[216] Leone, Gustavo & Nuckolls, Faison & Ishida, Seiichi & Adams, Monique & Sears, Rosalie & Jakoi, Laszlo & Miron, Alexander & Nevins, Joseph. (2000). Identification of a Novel E2F3 Product Suggests a Mechanism for Determining Specificity of Repression by Rb Proteins. Molecular and cellular biology. 20. 3626-32. 10.1128/MCB.20.10.3626-3632.2000.
[217] Black, Esther & Hallstrom, Timothy & Dressman, Holly & West, Mike & Nevins, Joseph. (2005). Distinctions in the specificity of E2F function revealed by gene expression signatures. Proceedings of the National Academy of Sciences of the United States of America. 102. 15948-53. 10.1073/pnas.0504300102.
[218] Danielian, P.S. & Friesenhahn, L.B. & Faust, Ann Marie & West, Julie & Caron, A.M. & Bronson, Roderick & Lees, J.A.. (2008). E2f3a and E2f3b make overlapping but different contributions to total E2f3 activity. Oncogene. 27. 6561-70. 10.1038/onc.2008.253.
[219] Rojas López, Maria & Monzón, Pablo & Acerenza, Luis. (2021). A model for the regulation of apoptosis intrinsic pathway. The potential role of the transcriptional regulator E2F in the point of no return. Journal of Theoretical Biology. 525. 110765. 10.1016/j.jtbi.2021.110765.
[220] Mcclellan, Kelly & Ruzhynsky, Vladimir & Douda, David & Vanderluit, Jacqueline & Ferguson, Kerry & Chen, Danian & Bremner, Rod & Park, David & Leone, Gustavo & Slack, Ruth. (2007). Unique Requirement for Rb/E2F3 in Neuronal Migration: Evidence for Cell Cycle-Independent Functions. Molecular and cellular biology. 27. 4825-43. 10.1128/MCB.02100-06.
[221] Field, Seth & Tsai, Fong-Ying & Kuo, Frank & Zubiaga, Ana & Kaelin, William & Livingston, David & Orkin, Stuart & Greenberg, Michael. (1996). E2F-1 Functions in Mice to Promote Apoptosis and Suppress Proliferation. Cell. 85. 549-61. 10.1016/S0092-8674(00)81255-6.
[222] Yamasaki, Lili & Jacks, Tyler & Bronson, Roderick & Goillot, Evelyne & Harlow, Ed & Dyson, Nicholas. (1996). Tumor Induction and Tissue Atrophy in Mice Lacking E2F-1. Cell. 85. 537-48. 10.1016/S0092-8674(00)81254-4.
[223] Humbert, Patrick & Rogers, Catherine & Ganiatsas, Soula & Landsberg, Rebecca & Trimarchi, Jeffrey & Dandapani, Savita & Brugnara, Carlo & Erdman, Susan & Schrenzel, Mark & Bronson, Roderick & Lees, Jacqueline. (2000). E2F4 Is Essential for Normal Erythrocyte Maturation and Neonatal Viability. Molecular cell. 6. 281-91. 10.1016/S1097-2765(00)00029-0.
[224] Yoshizuka, Naoto & New, Liguo & Moser, Bettina & Li, Yilei & Liao, Rong & Xie, Changchuan & Chen, Jianming & Deng, Qingdong & Martinez-Yamout, Maria & Dong, Meng-Qiu & Frangou, Costas & Yates, John & Wright, Peter & Han, Jiahuai. (2007). PRAK Is Essential for ras-Induced Senescence and Tumor Suppression. Cell. 128. 295-308. 10.1016/j.cell.2006.11.050.
[225] Meng, R & Phillips, P & El-Deiry, W. (1999). P53-independent increase in E2F-1 expression enhances the cytotoxic effects of etoposide and of adriamycin. International journal of oncology. 14. 5-14. 10.3892/ijo.14.1.5.
[226] Weei-Chin, L. & Lin, F.-T & Nevins, Joseph. (2011). Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev.. 15. 1833-1844.
[227] Zhu, Jing & Field, Seth & Gore, Lia & Thompson, Margaret & Yang, Haidi & Fujiwara, Yuko & Cardiff, Robert & Greenberg, Michael & Orkin, Stuart & Degregori, James. (2002). E2F1 and E2F2 Determine Thresholds for Antigen-Induced T-Cell Proliferation and Suppress Tumorigenesis. Molecular and cellular biology. 21. 8547-64. 10.1128/MCB.21.24.8547-8564.2001.
[228] Wu, Lizhao & Timmers, Cynthia & Maiti, Baidehi & Saavedra, Harold & Sang, Ling & Chong, Gabriel & Nuckolls, Faison & Giangrande, Paloma & Wright, Fred & Field, Seth & Greenberg, Michael & Orkin, Stuart. (2021). essential for cellular proliferation.
[229] Dyson, N & Dembski, M & Fattaey, A & Ngwu, C & Ewen, M & Helin, Kristian. (1994). Analysis of p107-associated proteins: p107 Associates with a form of E2F that differs from pRB-associated E2F-1. Journal of virology. 67. 7641-7. 10.1128/JVI.67.12.7641-7647.1993.
[230] Ikeda, Masa-Aki & Jakoi, L & Nevins, Joseph. (1996). Ikeda M-A, Jakoi L, Nevins JR.. A unique role for the Rb protein in controlling E2F accumulation during cell growth and differentiation. Proc Natl Acad Sci USA 93: 3215-3220. Proceedings of the National Academy of Sciences of the United States of America. 93. 3215-20. 10.1073/pnas.93.8.3215.
[231] Moberg, Ken & Starz, MA & Lees, JA. (1996). Moberg K, Starz MA, Lees JA.. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol Cell Biol 16: 1436-1449. Molecular and cellular biology. 16. 1436-49. 10.1128/MCB.16.4.143
[232] Muller, Heiko & Moroni, Cristina & Vigo, E. & Petersen, B. & Bartek, Jiri & Helin, Kristian. (2021). Induction of S-phase entry by E2F transcription Wu.
[233] Verona, R & Moberg, Ken & Estes, Scott & Starz, M & Vernon, J & Lees, J. (1998). E2F activity is regulated by cell cycle-dependent changes in subcellular localization. Molecular and cellular biology. 17. 7268-82. 10.1128/MCB.17.12.7268.
[234] Magae, Junji & Wu, Chin-Lee & Illenye, Sharon & Harlow, E & Heintz, N. (1996). Nuclear localization of DP and E2F transcription factors by heterodimeric partners and retinoblastoma protein family members. Journal of cell science. 109 ( Pt 7). 1717-26.
[235] Gaubatz, Stefan & Lees, Jacqueline & Lindeman, Geoffrey & Livingston, David. (2001). E2F4 Is Exported from the Nucleus in a CRM1-Dependent Manner. Molecular and cellular biology. 21. 1384-92. 10.1128/MCB.21.4.1384-1392.2001.
[236] Amerongen, Machteld & Diehl, Florian & Novoyatleva, Tatyana & Patra, Chinmoy & Engel, Felix. (2009). E2F4 is required for cardiomyocyte proliferation. Cardiovascular research. 86. 92-102. 10.1093/cvr/cvp383.
[237] Varone, Antonio & Massagué, Joan. (1999). E2F and histone deacetylase mediate transforming growth factor beta repression of cdc25A during keratinocyte cell cycle arrest. Molecular and cellular biology. 19. 916-22. 10.1128/MCB.19.1.916.
[238] Hsiao, Kuang-Ming & McMahon, S & Farnham, Peggy. (1994). Multiple DNA elements are required for the growth regulation of the mouse E2F1 promoter. Genes & development. 8. 1526-37. 10.1101/gad.8.13.1526.
[239] Taubert, Stefan & Gorrini, Chiara & Frank, Scott & Parisi, Tiziana & Fuchs, Miriam & Chan, Homan & Livingston, David & Amati, Bruno. (2004). E2F-Dependent Histone Acetylation and Recruitment of the Tip60 Acetyltransferase Complex to Chromatin in Late G1. Molecular and cellular biology. 24. 4546-56. 10.1128/MCB.24.10.4546-4556.2004.
[240] Catchpole, Steven & Tavner, Fiona & Cam, Laurent & Sardet, Claude & Watson, Roger. (2002). A B-myb Promoter Corepressor Site Facilitatesin Vivo Occupation of the Adjacent E2F Site by p107·E2F and p130·E2F Complexes. The Journal of biological chemistry. 277. 39015-24. 10.1074/jbc.M202960200.
[241] Bruce, Jacqueline & Hurford, Robert & Classon, Marie & Koh, James & Dyson, Nicholas. (2000). Requirements for Cell Cycle Arrest by p16INK4a. Molecular cell. 6. 737-42. 10.1016/S1097-2765(00)00072-1.
[242] Hurford, R.K. & Cobrinik, David & Lee, M & Dyson, N. (1997). Hurford Jr KR, Cobrinik D, Lee HM, Dyson N.. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev 11: 1447-1463. Genes & development. 11. 1447-63. 10.1101/gad.11.11.1447.
[243] Rempel, Rachel & Sáenz Robles, Maria & Storms, Robert & Morham, Scott & Ishida, Seiichi & Engel, Amber & Jakoi, Laszlo & Melhem, Mona & Pipas, James & Smith, Clay & Nevins, Joseph. (2000). Loss of E2F4 Activity Leads to Abnormal Development of Multiple Cellular Lineages. Molecular cell. 6. 293-306. 10.1016/S1097-2765(00)00030-7.
[244] Persengiev, Stephan & Kondova, Ivanela & Kilpatrick, Daniel. (1999). E2F4 Actively Promotes the Initiation and Maintenance of Nerve Growth Factor-Induced Cell Differentiation. Molecular and cellular biology. 19. 6048-56. 10.1128/MCB.19.9.6048.
[245] Cobrinik, David & Lee, M.H. & Hannon, G & Mulligan, George & Bronson, Roderick & Dyson, N & Harlow, E & Beach, David & Weinberg, R.A. & Jacks, T. (1996). Shared role of the pRB-related p130 and p107 proteins in limb development. Genes & development. 10. 1633-44. 10.1101/gad.10.13.1633.
[246] Porse, Bo & Pedersen, Thomas & Xu, Xiufeng & Lindberg, Bo & Wewer, Ulla & Friis-Hansen, Lennart & Nerlov, Claus. (2001). E2F Repression by C/EBPα Is Required for Adipogenesis and Granulopoiesis In Vivo. Cell. 107. 247-58. 10.1016/S0092-8674(01)00516-5.
[247] Chen, Phang-Lang & Riley, D & Chen-Kiang, S & Lee, W. (1996). Retinoblastoma protein directly interacts and activates the transcription factor NF-IL6. Proceedings of the National Academy of Sciences of the United States of America. 93. 465-9. 10.1073/pnas.93.1.465.
[248] Morkel, Markus & Wenkel, J & Bannister, Andrew & Kouzarides, T & Hagemeier, Christian. (1998). An E2F-like repressor of transcription. Nature. 390. 567-8. 10.1038/37507.
[249] Gaubatz, Stefan & Wood, Jason & Livingston, David. (1998). Unusual Proliferation Arrest and Transcriptional Control Properties of a Newly Discovered E2F Family Member, E2F-6. Proceedings of the National Academy of Sciences of the United States of America. 95. 9190-5. 10.1073/pnas.95.16.919
[250] Trimarchi, Jeffrey & Fairchild, Brian & Wen, Jessica & Lees, Jacqueline. (2001). The E2F6 transcription factor is a component of the mammalian Bmi1-containing polycomb complex. Proceedings of the National Academy of Sciences of the United States of America. 98. 1519-24. 10.1073/pnas.041597698.
[251] Francis, Nicole & Kingston, Robert. (2001). Mechanisms of transcriptional memory. Nature reviews. Molecular cell biology. 2. 409-21. 10.1038/35073039
[252] Relaix, Frédéric & Weng, Xun & Marazzi, Giovanna & Yang, Ellen & Copeland, Neal & Jenkins, Nancy & Spence, Sally & Sassoon, David. (1996). Pw1, a Novel Zinc Finger Gene Implicated in the Myogenic and Neuronal Lineages. Developmental biology. 177. 383-96. 10.1006/dbio.1996.0172.
[253] Jacobs, Jacqueline & Scheijen, Blanca & Voncken, Jw & Kieboom, Karin & Berns, A. & Lohuizen, Maarten. (1999). Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes & development. 13. 2678-90. 10.1101/gad.13.20.2678.
[254] Dynlacht, B & Brook, Adam & Dembski, M & Yenush, Lynne & Dyson, N. (1994). DNA-binding and trans-activation properties of Drosophila E2F and DP proteins. Proceedings of the National Academy of Sciences of the United States of America. 91. 6359-63. 10.1073/pnas.91.14.6359.
[255] Watson, J. & Crick, F.. (2017). A Structure for Deoxyribose Nucleic Acid. 10.1007/978-3-662-47150-0_4.
[256] https://www.nature.com/collections/mvrzbllfvv
[257] Watson, James & Crick, Francis. (2007). Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid.. Clinical orthopaedics and related research. 462. 3-5. 10.1097/BLO.0b013e31814b9304.
[258] Bayliss, M.. (1985). The cell division cycle in plants. SEB Seminar series 26.
[259] Taylor, J. & Woods, Philip & Hughes, Walter. (1957). The Organization and Duplication of Chromosomes as Revealed by Autoradiographic Studies Using Tritium Labeled Thymidine. Proceedings of the National Academy of Sciences of the United States of America. 43. 122-8. 10.1073/pnas.43.1.122.
[260] Meselson, Matthew & Stahl, Franklin. (1958). The replication of DNA in Escherichia-Coli. Proceedings of the National Academy of Sciences of the United States of America. 44. 671-82.
[261] Theis, James & Newlon, Carol. (1997). The ARS309 chromosomal replicator of Saccharomyces cerevisiae depends on an exceptional ARS consensus sequence. Proceedings of the National Academy of Sciences of the United States of America. 94. 10786-91. 10.1073/pnas.94.20.10786.
[262] Bielinsky, Anja & Gerbi, Susan. (2001). Where it all starts: Eukaryotic origins of DNA replication. Journal of cell science. 114. 643-51. 10.1242/jcs.114.4.643.
[263] Diffley, John & Cocker, Julie. (1992). Protein-DNA interactions at a yeast replication origin. Nature. 357. 169-72. 10.1038/357169a0.
[264] Crouzet, Joel y Soubrier, Fabienne. Molécula de ADN circular con un origen de replicación condicional, su procedimiento de preparación y su utilización en terapia génica. Oficina Española de Patentes y Marcas. Patente Prioridad: 15.09.1995 FR 95 10825 (2005)
[265] Cadoret, Jean-Charles & Meisch, Françoise & Hassan-Zadeh, Vahideh & Luyten, Isabelle & Guillet, Claire & Duret, Laurent & Quesneville, Hadi & Prioleau, Marie-Noëlle. (2008). Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proceedings of the National Academy of Sciences of the United States of America. 105. 15837-42. 10.1073/pnas.0805208105
[266] Weinert, T.A. & Hartwell, L.H.. (1988). Weinert, T. A. & Hartwell, L. H. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 241, 317-322. Science (New York, N.Y.). 241. 317-22. 10.1126/science.3291120.
[267] Levine, Arnold. (1997). p53, the Cellular Gatekeeper for Growth and Division. Cell. 88. 323-31. 10.1016/S0092-8674(00)81871-1.
[268] Thakker, Nalin. (1993). Medical genetics: advances in brief: A mammalian cell cycle checkpoint utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Journal of Medical Genetics. 30. 10.1136/jmg.30.4.349-d.
[269] El-Deiry, Wafik & Kern, Scott & Pietenpol, Jennifer & Kinzler, Kenneth & Vogelstein, Bert. (1992). Definition of a consensus binding site for p53. Nature genetics. 1. 45-9. 10.1038/ng0492-45.
[270] Meng, Raymond & El-Deiry, Wafik. (2001). Meng, R. D. & el-Deiry, W. S. p53-independent upregulation of KILLER/DR5 TRAIL receptor expression by glucocorticoids and interferon-. Exp. Cell Res. 262, 154-169. Experimental cell research. 262. 154-69. 10.1006/excr.2000.5073.
[271] Helin, Kristian & Wu, Chin-Lee & Fattaey, AR & Lees, JA & Dynlacht, B & Ngwu, C & Harlow, E. (1993). Heterodimerization of the transcription factors E2F-1 and DP-1 leads to cooperative trans-activation. Genes & development. 7. 1850-61. 10.1101/gad.7.10.1850.
[272] Knudsen, Karen & Booth, Dana & Naderi, Soheil & Chroneos, Zvjezdana & Fribourg, Anne & Hunton, Irina & Feramisco, James & Wang, Jean & Knudsen, Erik. (2000). RB-dependent S-phase response to DNA damage. Molecular and cellular biology. 20. 7751-63. 10.1128/MCB.20.20.7751-7763.2000.
[273] Lindahl, Tomas. (1993). Instability and Decay of the Primary Structure of DNA. Nature. 362. 709-15. 10.1038/362709a0.
[274] Bebenek, Anna. (2008). [DNA replication fidelity].. Postepy biochemii. 54. 43-56. 10.1074/jbc.R400006200.
[275] Bootsma, Dirk & Hoeijmakers, Jan. (1996). DNA repair and mutagenesis. Trends in Genetics - TRENDS GENET. 12. 79-79. 10.1016/0168-9525(96)81408-9.
[276] Friedberg, E. (2000). Biological Responses to DNA Damage: A Perspective in the New Millennium. Cold Spring Harbor symposia on quantitative biology. 65. 593-602. 10.1101/sqb.2000.65.593.
[277] Lindahl, Tomas. (2000). Suppression of spontaneous mutagenesis in human cells by DNA base excision-repair. Mutation research. 462. 129-35. 10.1016/S1383-5742(00)00024-7.
[278] Buermeyer, Andrew & Deschenes, Suzanne & Baker, Sean & Liskay, R Michael. (1999). Mammalian DNA mismatch repair. Annual review of genetics. 33. 533-64. 10.1146/annurev.genet.33.1.533.
[279] Wood, Richard. (1997). Nucleotide Excision Repair in Mammalian Cells. The Journal of biological chemistry. 272. 23465-8. 10.1074/jbc.272.38.23465.
[280] Prakash, Shamsher & Prakash, Louise. (2000). Nucleotide excision repair in yeast. Mutation research. 451. 13-24. 10.1016/S0027-5107(00)00037-3.
[281] Svejstrup, Jesper & Wang, Zhigang & Feaver, W & Wu, Xiahua & Bushnell, David & Donahue, Thomas & Friedberg, Errol & Kornberg, Roger. (1995). Different forms of TFIIH for transcription and DNA repair. Cell. 80. 21-8. 10.1016/0092-8674(95)90447-6.
[282] Feaver, William. (2000). Subunit interactions in yeast transcription/repair factor TFIIH. Requirement for Tfb3 subunit in nucleotide excision repair. Journal of Biological Chemistry. 275. 5941-5946. 10.1074/jbc.275.8.5941.
[283] Kemp, Michael & Sancar, Aziz. (2012). DNA excision repair. Cell cycle (Georgetown, Tex.). 11. 2997-3002. 10.4161/cc.21126.
[284] Sugasawa, Kaoru & Okamoto, Tomoko & Shimizu, Yuichiro & Masutani, Chikahide & Iwai, Shigenori & Hanaoka, Fumio. (2001). A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes & development. 15. 507-21. 10.1101/gad.866301.
[285] Neilson, C. & Santos-Rosa, H. & Bannister, Andrew & Kouzarides, T.. (2006). Chromatin modifications and their function. 22-22.
[286] Krasikova, Y.S. & Rechkunova, N & Maltseva, Ekaterina & Craescu, C & Petruseva, Irina & Lavrik, Olga. (2012). Influence of centrin 2 on the interaction of nucleotide excision repair factors with damaged DNA. Biochemistry. Biokhimii?a. 77. 346-53. 10.1134/S0006297912040050.
[287] Vasquez, Karen & Christensen, Jesper & Li, Lei & Finch, Rick & Glazer, Peter. (2002). Human XPA and RPA DNA repair proteins participate in specific recognition of triplex-induced helical distortions. Proceedings of the National Academy of Sciences of the United States of America. 99. 5848-53. 10.1073/pnas.082193799.
[288] Sugasawa, Kaoru & Okuda, Yuki & Saijo, Masafumi & Nishi, Ryotaro & Matsuda, Noriyuki & Chu, Gilbert & Mori, Toshio & Iwai, Shigenori & Tanaka, Keiji & Tanaka, Kiyoji & Hanaoka, Fumio. (2005). UV-Induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex.. Cell. 121. 387-400. 10.1016/j.cell.2005.02.035.
[289] Lee, Jung-Hoon & Lee, Changwook & Yi, Ki & Cho, Yunje. (2005). Structure of a peptide:N-glycanase-Rad23 complex: Insight into the deglycosylation for denatured glycoproteins. Proceedings of the National Academy of Sciences of the United States of America. 102. 9144-9. 10.1073/pnas.0502082102.
[290] Sugasawa, Kaoru & Masutani, C & Uchida, A & Maekawa, T & Van der Spek, Peter & Bootsma, D & Hoeijmakers, J. & Hanaoka, Fumio. (1996). HHR23B, a human RAD23 homolog, stimulates XPC protein in nucleotide excision repair in vitro. Molecular and cellular biology. 16. 4852-61. 10.1128/MCB.16.9.4852.
[291] Araki, Marito & Masutani, Chikahide & Takemura, Mitsuyo & Uchida, Akio & Sugasawa, Kaoru & Kondoh, Jun & Ohkuma, Yoshiaki & Hanaoka, Fumio. (2001). Centrosome Protein Centrin 2/Caltractin 1 Is Part of the Xeroderma Pigmentosum Group C Complex That Initiates Global Genome Nucleotide Excision Repair. The Journal of biological chemistry. 276. 18665-72. 10.1074/jbc.M100855200.
[292] Hess, Martin & Schwitter, Urs & Petretta, Mario & Giese, Bernd & Naegeli, Hanspeter. (1997). Bipartite substrate discrimination by human nucleotide excision repair. Proceedings of the National Academy of Sciences of the United States of America. 94. 6664-9. 10.1073/pnas.94.13.6664.
[293] Sánchez, Tania & Hernández, Obdulio. (2010). El receptor del factor de crecimiento epidérmico y su papel en el desarrollo tumoral. Revista Habanera de Ciencias Médicas. 9. 172-180.
[294] Yoder, Kristine & Sarasin, Alain & Kraemer, Kenneth & Mcilhatton, Michael & Bushman, Frederic & Fishel, Richard. (2006). The DNA repair genes XPB and XPD defend cells from retroviral infection. Proceedings of the National Academy of Sciences of the United States of America. 103. 4622-7. 10.1073/pnas.0509828103.
[295] Merino, Carlos & Reynaud, Enrique & Vázquez, Martha & Zurta, Mario. (2002). DNA Repair and Transcriptional Effects of Mutations in TFIIH in Drosophila Development. Molecular biology of the cell. 13. 3246-56. 10.1091/mbc.E02-02-0087.
[296] Mu, David & Park, Chi-Hyun & Matsunaga, Tsukasa & Hsu, David & Reardon, Joyce & Sancar, Aziz. (1995). Reconstitution of Human DNA Repair Excision Nuclease in a Highly Defined System. The Journal of biological chemistry. 270. 2415-8. 10.1074/jbc.270.6.2415.
[297] Wakasugi, Mitsuo & Sancar, Aziz. (1999). Order of Assembly of Human DNA Repair Excision Nuclease. The Journal of biological chemistry. 274. 18759-68. 10.1074/jbc.274.26.18759.
[298] Cotter, Laura & Allen, Terence & Kiseleva, Elena & Goldberg, Martin. (2007). Nuclear Membrane Disassembly and Rupture. Journal of molecular biology. 369. 683-95. 10.1016/j.jmb.2007.03.051.
[299] Wood, R.D.. (1996). Human DNA repair endonuclease subunit (XPF) mRNA, complete cds.
[300] Carpenter, Edward. (1985). Book Review:Dinoflagellates. David L. Spector. Quarterly Review of Biology - QUART REV BIOL. 60. 10.1086/414615.
[301] Haaf, Thomas & Hayman, David. (1991). Quantitative determination of rDNA transcription units in vertebrate cells. Experimental cell research. 193. 78-86. 10.1016/0014-4827(91)90540-B.
[302] Manuelidis, Laura & Borden, Jonathan. (1988). Reproducible compartmentalization of individual chromosome domains in human CNS cells revealed by in situ hybridization and 3-D reconstruction. Chromosoma. 96. 397-410. 10.1007/BF00303033.
[303] Vourc'h, Claire & Taruscio, Domenica & Boyle, Ann & Ward, David. (1993). Cell Cycle-Dependent Distribution of Telomeres, Centromeres, and Chromosome-Specific Subsatellite Domains in the Interphase Nucleus of Mouse Lymphocytes. Experimental cell research. 205. 142-51. 10.1006/excr.1993.1068.
[304] Popp, Susanne & Scholl, Hans & Loos, Peter & Jauch, Anna & Stelzer, Ernst & Cremer, Christoph & Cremer, Thomas. (1990). Distribution of chromosome 18 and X centric heterochromatin in the interphase nucleus of cultured human cells. Experimental Cell Research. 189. 1-12. 10.1016/0014-4827(90)90249-A.
[305] Bridger, Joanna & Herrmann, Harald & Münkel, C & Lichter, P.J.. (1998). Identification of an interchromosomal compartment by polymerization of nuclear-targeted vimentin. Journal of cell science. 111 ( Pt 9). 1241-53.
[306] Hofmeister, W.. (2021). Zusatze und Berichtigungen zu den 1851 veröffentlichen Untersuchungengen der Entwicklung höherer Kryptogamen. Jahrbucher für Wissenschaft und Botanik. 3. 259-293.
[307] Lynch, Timothy & Lintilhac, Philip. (1997). Mechanical Signals in Plant Development: A New Method for Single Cell Studies. Developmental biology. 181. 246-56. 10.1006/dbio.1996.8462.
[308] Pickett-Heaps, J & Northcote, D. (1966). Organization of Microtubules and Endoplasmic Reticulum During Mitosis and Cytokinesis in Wheat Meristems. Journal of cell science. 1. 109-20. 10.1242/jcs.1.1.109.
[309] Staehelin, L. & Hepler, Peter. (1996). Cytokinesis in Higher Plants. Cell. 84. 821-4. 10.1016/S0092-8674(00)81060-0.
[310] Monteiro, M.C. & O´Connor, J.E. & Martínez, M.. (2001). La Citometría de Flujo en el Análisis de las Plaquetas: (I) Aspectos Estructurales y Funcionales de las Plaquetas. Revista de Diagnostico Biologico. 50. 111-136.
[311] Dokland, Terje & Mckenna, Robert & Ilag, Leodevico & Bowman, Brian & Incardona, Nino & Fane, Bentley & Rossmann, Michael. (1997). Structure of a viral procapsid with molecular scaffolding. Nature. 389. 308-13. 10.1038/38537.
[312] Yvon, Anne-Marie & Walker, Jonathan & Danowski, Barbara & Fagerstrom, Carey & Khodjakov, Alexey & Wadsworth, Pat. (2002). Centrosome Reorientation in Wound-Edge Cells Is Cell Type Specific. Molecular biology of the cell. 13. 1871-80. 10.1091/mbc.01-11-0539.
[313] DeBonis, Salvatore & Simorre, Jean-Pierre & Crevel, Isabelle & Lebeau, Luc & Skoufias, Dimitrios & Blangy, Anne & Ebel, Christine & Gans, Pierre & Cross, Robert & Hackney, David & Wade, Richard & Kozielski, Frank. (2003). Interaction of the Mitotic Inhibitor Monastrol with Human Kinesin Eg5 †. Biochemistry. 42. 338-49. 10.1021/bi026716j.
[314] Presley, John & Smith, Carolyn & Hirschberg, Koty & Miller, Chad & Cole, Nelson & Zaal, Kristien & Lippincott-Schwartz, Jennifer. (1998). Golgi Membrane Dynamics. Molecular biology of the cell. 9. 1617-26. 10.1091/mbc.9.7.1617.
[315] Rietdorf, Jens & Ploubidou, Aspasia & Reckmann, Inge & Holmström, Anna & Frischknecht, Friedrich & Zettl, Markus & Zimmermann, Timo & Way, Michael. (2001). Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nature cell biology. 3. 992-1000. 10.1038/ncb1101-992.
[316] Cytrynbaum, Eric & Scholey, J & Mogilner, A. (2003). A Force Balance Model of Early Spindle Pole Separation in Drosophila Embryos. Biophysical journal. 84. 757-69. 10.1016/S0006-3495(03)74895-4.
[317] Granzier, Henk & Helmes, Michiel & Trombitás, K. (1996). Nonuniform elasticity of titin in cardiac myocytes: A study using immunoelectron microscopy and cellular mechanics. Biophysical journal. 70. 430-42. 10.1016/S0006-3495(96)79586-3.
[318] Wilson, J & Biden, T & Ludowyke, R. (2000). Increases in phosphorylation of the myosin II heavy chain but not regulatory light chains correlate with insulin secretion in rat pancreatic islets and RINm5F cells. Diabetes. 48. 2383-9. 10.2337/diabetes.48.12.2383.
[319] Bandyopadhyay, Sanghamitra & Ashok, Anushruti & Rai, Nagendra & Rai, Asit & Tripathi, Sachin. (2014). Model of Alzheimer’s disease.
[320] West, Robert & Malmstrom, Terra & McIntosh, Richard. (2002). Kinesins klp5(+) and klp6(+) are required for normal chromosome movement in mitosis. Journal of cell science. 115. 931-40. 10.1242/jcs.115.5.931.
[321] Desai, Arshad & Deacon, Heather & Walczak, Claire & Mitchison, Timothy. (1997). A method that allows the assembly of kinetochore components onto chromosomes condensed in clarified Xenopus egg extracts. Proceedings of the National Academy of Sciences of the United States of America. 94. 12378-83. 10.1073/pnas.94.23.12378.
[322] Putkey, Frances & Cramer, Thorsten & Morphew, Mary & Silk, Alain & Johnson, Randall & McIntosh, Richard & Cleveland, Don. (2002). Unstable Kinetochore-Microtubule Capture and Chromosomal Instability Following Deletion of CENP-E. Developmental cell. 3. 351-65. 10.1016/S1534-5807(02)00255-1.
[323] Abrieu, Ariane & Kahana, Jason & Wood, Kenneth & Cleveland, Don. (2000). CENP-E as an Essential Component of the Mitotic Checkpoint In Vitro. Cell. 102. 817-26. 10.1016/S0092-8674(00)00070-2.
[324] Mitchison, Timothy & Salmon, E.D.. (2001). Mitosis: A history of division. Nature cell biology. 3. E17-21. 10.1038/35050656.
[325] Funabiki, Hironori & Murray, Andrew. (2000). The Xenopus Chromokinesin Xkid Is Essential for Metaphase Chromosome Alignment and Must Be Degraded to Allow Anaphase Chromosome Movement. Cell. 102. 411-24. 10.1016/S0092-8674(00)00047-7.
[326] Ai, Bao-Quan & Xian-Ju, Wang & Liu, Lianggang & Nakano, Masahiro & Matsuura, H.. (2001). A Microscopic Mechanism for Muscle Motion. Chinese Physics Letters. 19. 137. 10.1088/0256-307X/19/1/343.
[327] Meyer, Gary & Feldman, Eva. (2002). Signaling mechanisms that regulate actin-based motility processes in the nervous system. Journal of neurochemistry. 83. 490-503. 10.1046/j.1471-4159.2002.01185.x.
[328] Astumian, Raymond & Derényi, Imre. (1998). Fluctuation driven transport and models of molecular motors and pumps. European biophysics journal : EBJ. 27. 474-89. 10.1007/s002490050158.
[329] Martinell, Julio & Raga, A. & Velazquez, Pablo. (2003). Effects of Anisotropic Thermal Conduction on X-Ray Emission from SNRS. Revista Mexicana de Astronomía y Astrofísica. 15.
[330] Stracke, Roland & Böhm, Konrad & Burgold, Jörg & Schacht, Hans-Joachim & Unger, Eberhard. (1999). Physical and technical parameters determining kinesin-driven microtubule motility in vitro.
[331] Rogers, Stephen & Karcher, Ryan & Roland, Joseph & Minin, Alexander & Steffen, Walter & Gelfand, Vladimir. (1999). Regulation of Melanosome Movement in the Cell Cycle by Reversible Association with Myosin V. The Journal of cell biology. 146. 1265-76. 10.1083/jcb.146.6.1265.
[332] Reimann, Peter & Elston, Timothy. (1997). Kramers Rate for Thermal Plus Dichotomous Noise Applied to Ratchets. Physical review letters. 77. 5328-5331. 10.1103/PhysRevLett.77.5328.
[333] Wang, Xiao & Hao, Nan & Dohlman, Henrik & Elston, Timothy. (2006). Bistability, Stochasticity, and Oscillations in the Mitogen-Activated Protein Kinase Cascade. Biophysical journal. 90. 1961-78. 10.1529/biophysj.105.073874.
[334] LaFountain, James & Cole, Richard & Rieder, Conly. (2002). Partner telomeres during anaphase in crane-fly spermatocytes are connected by an elastic tether that exerts a backward force and resists poleward motion. Journal of cell science. 115. 1541-9.
[335] Nicklas, R. & Waters, Jennifer & Salmon, E & Ward, Suzanne. (2002). Checkpoint signals in grasshopper meiosis are sensitive to microtubule attachment, but tension is still essential. Journal of cell science. 114. 4173-83. 10.1242/jcs.114.23.4173.
[336] Taylor, Stephen & Hussein, Deema & Wang, YM & Elderkin, Sarah & Morrow, Christopher. (2002). Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. Journal of cell science. 114. 4385-95. 10.1242/jcs.114.24.4385.
[337] Mcewen, Bruce & Chan, Gordon & Zubrowski, Beata & Savoian, Matthew & Sauer, Matthew & Yen, Tim. (2001). CENP-E Is Essential for Reliable Bioriented Spindle Attachment, but Chromosome Alignment Can Be Achieved via Redundant Mechanisms in Mammalian Cells. Molecular biology of the cell. 12. 2776-89. 10.1091/mbc.12.9.2776.
[338] Williams, Byron & Li, ZeXiao & Liu, Song-tao & Williams, Erika & Leung, Garmay & Yen, Tim & Goldberg, Michael. (2003). Zwilch, a New Component of the ZW10/ROD Complex Required for Kinetochore Functions. Molecular biology of the cell. 14. 1379-91. 10.1091/mbc.E02-09-0624.
[339] Banks, Jennifer & Heald, Rebecca. (2001). Chromosome movement: Dynein-out at the kinetochore. Current biology : CB. 11. R128-31. 10.1016/S0960-9822(01)00059-8.
[340] Ruvolo, P. (2001). Ceramide regulates cellular homeostasis via diverse stress signaling pathways. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U.K. 15. 1153-60. 10.1038/sj.leu.2402197.