Texto de apropiación científica y tecnológica_____________________________
Módulo 4. Quimioterapia viral
4.1 Quimioterapia
4.2 Estrategias para el desarrollo de agentes antivirales
4.3 Aplicación clínica
4.3.1 Formulación y métodos de administración
4.4 Estrategias clínicas
4.5 Prevención de la transmisión de virus
4.6 Mecanismos de acción y papel de los antivirales individualmente
4.6.1 Interferones
4.6.2 Bloqueo de la adhesión o fusión
4.6.2 Bloqueo del revestimiento: bloqueadores de canales iónicos
4.6.3 Inhibidores de virus de ARN
4.6.4 Inhibidores de la transcriptasa inversa
4.6.5 Inhibidores de la transcriptasa inversa no nucleósidos
4.6.6 Inhibidores de proteasas virales
4.6.7 Inhibidores de neuraminidasa
____________________________________________________________________________
3.1 Quimioterapia
En las últimas dos décadas, la terapia antiviral ha experimentado una revolución. Después de una larga era de esperanza atenuada por la decepción cuando solo había uno o dos agentes exitosos, muchas infecciones virales ahora pueden tratarse con uno o más regímenes antivirales, y nuestra comprensión de las estrategias quimioterapéuticas y la resistencia a los medicamentos se ha vuelto considerablemente más sofisticada. La mayoría de los agentes de la Tabla 4.1 se han desarrollado en los últimos 20 años (Recuadro 4.1).

En términos generales, hay tres clases de agentes químicos que se usan para combatir infecciones:
1. Los medicamentos antivirales actúan suprimiendo o previniendo la replicación viral en las células infectadas, y son lo suficientemente no tóxicos como para que puedan usarse para tratar pacientes infectados.
2. Los desinfectantes están diseñados para destruir la infectividad de las partículas virales libres, pero en su mayoría son tóxicos para la célula huésped y no tienen un efecto clínico significativo contra la infección establecida.
3. Los muchos antibióticos disponibles ahora para combatir las bacterias no tienen actividad contra los virus. Las únicas circunstancias en las que puede ser apropiado prescribir antibióticos en infecciones virales son (1) para prevenir o tratar una sobreinfección bacteriana grave de una enfermedad viral (p. ej., neumonía bacteriana que complica la influenza), o (2) para ir a lo seguro cuando existe una posibilidad real de una enfermedad bacteriana grave, como meningitis o neumonía, hasta que se disponga de la identificación de laboratorio del agente etiológico. Sin embargo, la práctica generalizada de prescribir antibióticos como respuesta instintiva a cualquier infección conlleva una serie de consecuencias indeseables y es una práctica médica irresponsable. El desarrollo de medicamentos antivirales se ha retrasado con respecto al éxito de los agentes antibacterianos por varias razones (Cuadro 4.2).

Sin embargo, tres grandes avances en las décadas de 1980 y 1990 proporcionaron a la industria farmacéutica un optimismo considerable. Estos fueron (1) el descubrimiento y el aumento del uso de aciclovir, (2) la producción de interferón mediante tecnología de ADN recombinante y (3) la introducción de una variedad cada vez mayor de medicamentos efectivos contra el VIH junto con una nueva comprensión de cómo estas armas pueden aplicarse en la práctica clínica. Ahora estamos viendo una gama de nuevos agentes contra muchas enfermedades virales humanas importantes que llegan a la etapa de ensayos clínicos, combinados con un conocimiento en gran expansión sobre cómo manejar dichos tratamientos y los efectos secundarios asociados. Cabe señalar que muchos de los fármacos antivirales actualmente disponibles son para el tratamiento de infecciones crónicas, donde el momento de la administración del fármaco no es tan crítico como para el tratamiento de infecciones agudas. Además, la mayoría de los fármacos solo suprimen la replicación, pero no erradican por sí mismos la infección, y el verdadero éxito terapéutico y la erradicación de la infección también requieren una respuesta inmunitaria eficaz; de lo contrario, se puede observar un rebote de la replicación cuando cesa la administración del fármaco.
4.2 Estrategias para el desarrollo de agentes antivirales
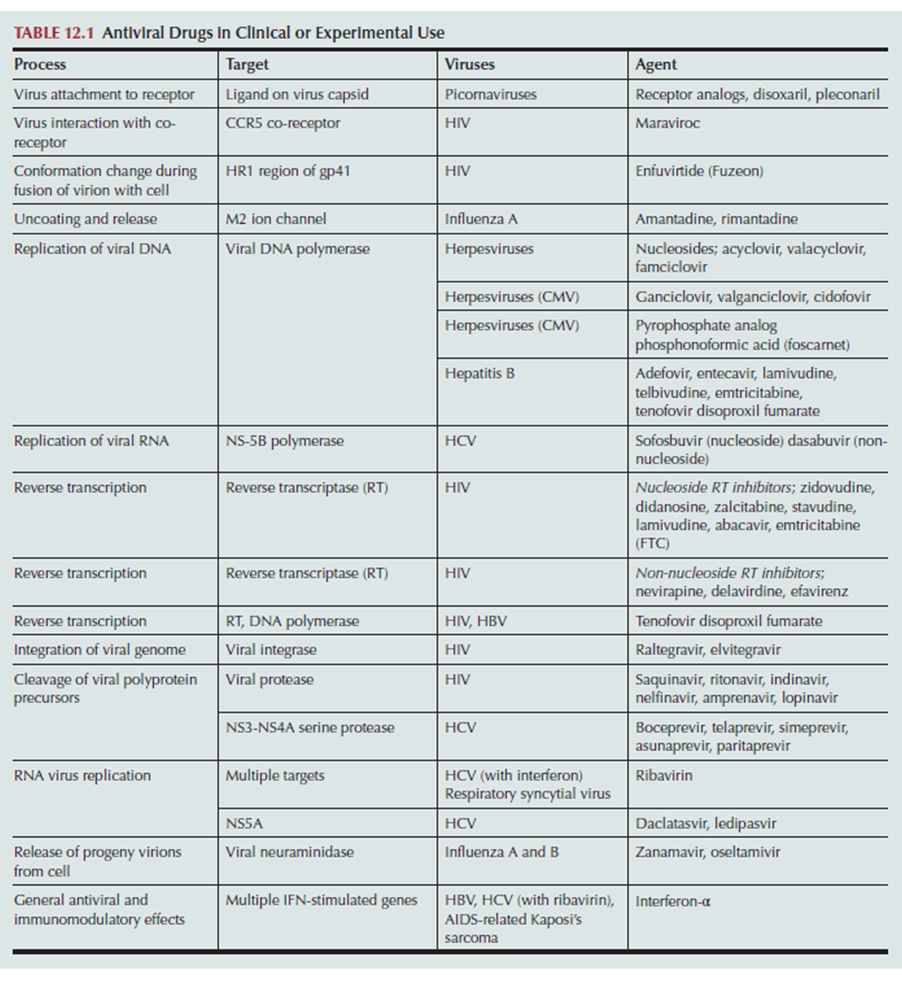
En teoría, cualquiera de los pasos en el ciclo de replicación viral representa posibles objetivos para el ataque quimioterapéutico selectivo. Los pasos exclusivos de la replicación del virus son objetivos potenciales, al igual que aquellos para los que se puede diseñar un inhibidor que tenga una mayor actividad selectiva contra un proceso específico del virus que contra un proceso huésped correspondiente. Los ejemplos se muestran en la Tabla 4.1.
TABLA 4.1 Medicamentos antivirales en uso clínico o experimental
Un refinamiento adicional de este enfoque se ilustra con el análogo de nucleósido acicloguanosina (aciclovir), que inhibe preferentemente la ADN polimerasa del herpesvirus esencial para la replicación del ADN viral, en lugar de las polimerasas de la célula huésped. De hecho, el aciclovir es un profármaco inactivo que requiere otra enzima codificada por el virus del herpes, la timidina quinasa, para fosforilarlo a su forma activa. Como la enzima vírica se produce únicamente en las células infectadas, estos profármacos no son tóxicos para las células no infectadas; la especificidad de su acción contra la replicación viral en lugar de la del huésped se basa en dos pasos selectivos separados.
Los nuevos agentes quimioterapéuticos antivirales pueden provenir de varias fuentes diferentes:
1. Las grandes compañías farmacéuticas albergan grandes bancos de compuestos con posibles actividades biológicas. Estos se seleccionan para una función deseada, por ejemplo, la inhibición de la replicación in vitro de un virus en particular, ya sea mediante pruebas directas de un fármaco candidato o como parte de protocolos de detección sofisticados de muy alto rendimiento. Un ejemplo es la AZT (zidovudina), el primer inhibidor de la transcriptasa inversa análogo de los nucleósidos (INTI) utilizado contra el VIH. Esto se sintetizó en 1964 como un posible fármaco contra el cáncer, pero se descubrió que no era efectivo. Mucho más tarde se informó que tenía actividad anti-retrovirus en 1974, y luego se examinó y se encontró que era activo contra el VIH a mediados de la década de 1980 (Cuadro 12.3). 2. Selección o síntesis de compuestos conocidos o previstos para inhibir un proceso objetivo en la replicación de virus, por ejemplo, mediante la unión a un sitio activo o intermediario 3. Modificación de compuestos que ya se sabe que tienen efectos antivirales ("clientes potenciales"), para mejorar la eficacia o superar los inconvenientes 4. Sustancias o productos naturales que se ha informado que tienen un beneficio terapéutico.
2. El conocimiento de la estructura detallada de un sitio activo crítico para la replicación del virus, por ejemplo, una enzima viral, un ligando para un receptor o un objetivo regulador, permite la creación de inhibidores específicos mediante un diseño. Los inhibidores de la neuraminidasa, una clase de inhibidores potentes de la replicación de la influenza, fueron desarrollados por Mark von Itzstein después de que Peter Colman resolviera la estructura fina de la proteína neuraminidasa mediante cristalografía de rayos X; este conocimiento permitió la síntesis racional de derivados químicos que se unen fuertemente al sitio activo de la enzima. Se puede utilizar un enfoque similar para desarrollar fármacos que actúen uniéndose directamente a la cápside (o envoltura) del propio virión, bloqueando así los primeros pasos del ciclo de replicación, por ejemplo, la unión, la penetración o el desprendimiento. Una vez que se conoce la estructura tridimensional de toda la superficie de los viriones isométricos, como los picornavirus, el sitio de unión al receptor (el ligando) en la proteína crítica de la cápside se puede caracterizar en detalle atómico. Los complejos de proteínas virales con receptores celulares purificados o imitadores de receptores pueden cristalizarse y examinarse directamente. El sitio de unión al receptor en el virión generalmente ha resultado ser un “cañón”, hendidura o depresión en la superficie externa de la proteína. Se toman medidas adicionales para analizar la estructura de la proteína viral después de unirse a un compuesto conocido por neutralizar la infectividad, para mapear los residuos de aminoácidos particulares que se encuentran sustituidos en mutantes resistentes del virus, o para usar mutagénesis específica del sitio para identificar residuos críticos. Luego, esta información se puede explotar para diseñar mejores drogas sintéticas, utilizando modelos informáticos para optimizar el ajuste y la energía de unión de la interacción fármaco-virus.
3. Una vez que se encuentra un agente que muestra un grado de inhibición específica para una enzima viral u otro proceso integral para la replicación viral (molécula "principal"), se sintetizan análogos relacionados (congéneres) del prototipo con miras a mejorar su actividad, solubilidad y biodisponibilidad, o reduciendo su toxicidad.
4. A menudo se propone que los productos naturales (p. ej., "medicamentos populares") que se reportan como beneficiosos contra enfermedades particulares pueden contener ingredientes activos a partir de los cuales se pueden desarrollar nuevas clases de antivirales potentes. Esta área de descubrimiento sigue siendo esperanzadora, pero con demasiada frecuencia estos productos resultan ser mezclas complejas y el aparente beneficio terapéutico parece desvanecerse a medida que el material se fracciona en sus componentes.
Un nuevo medicamento antiviral exitoso generalmente pasa por tres etapas de evaluación: detección in vitro inicial en cultivo celular para determinar su índice terapéutico contra un virus en particular, seguido de estudios en animales para determinar la farmacocinética, la toxicidad y (si existe un modelo adecuado) eficacia, seguida finalmente por sucesivos estudios de fase I, II y III en humanos. Para cualquier medicamento exitoso, este proceso puede llevar 10 años o más y puede costar muchos millones de dólares. Por lo general, muchos miles de otros candidatos potenciales deben descartarse a lo largo de esta ruta, después de haber fallado en una de las etapas sucesivas enumeradas en el Cuadro 4.4. La primera prueba clásica de cualquier agente quimioterapéutico putativo es, por supuesto, la inhibición de la replicación viral. En presencia de diluciones del agente, la multiplicación de La toxicidad del fármaco para las células humanas no infectadas puede medirse crudamente por el efecto citopático o, de forma más sensible, por la reducción de la eficacia del cultivo celular o el tiempo de duplicación celular. En general, solo vale la pena seguir adelante con aquellos agentes que muestran un índice terapéutico de al menos 10 y preferiblemente de 100 a 1000 (Cuadro 4.4).

Idealmente, el fármaco debe ser soluble en agua, química y metabólicamente estable, moderadamente apolar y absorbido satisfactoriamente por las células. Los estudios farmacocinéticos, primero en animales y luego en humanos, abordan cuestiones como el mecanismo y la velocidad de absorción siguiendo varias vías de administración, distribución tisular, metabolismo, desintoxicación y excreción del fármaco. Las pruebas de toxicidad aguda abarcan la vigilancia clínica integral de todos los sistemas del cuerpo, pruebas bioquímicas (por ejemplo, para la función hepática y renal), hematología, pruebas de inmunosupresión, etc. Las investigaciones a más largo plazo detectan la toxicidad crónica, la alergenicidad, la mutagenicidad, la carcinogenicidad y la teratogenicidad.
4.3 Aplicación clínica
4.3.1 Formulación y métodos de administración
La vía de administración de un agente antiviral es una consideración primordial al evaluar su aceptabilidad general. La vía oral es, naturalmente, con mucho la más conveniente para el paciente. Las gotas o aerosoles nasales pueden ser aceptables para las infecciones de las vías respiratorias superiores, pero pueden ser irritantes, mientras que la administración continua de aerosoles a través de una máscara facial o una tienda de oxígeno generalmente es apropiada solo para pacientes muy enfermos y hospitalizados. Las preparaciones tópicas (cremas, ungüentos, etc.) son satisfactorias para las infecciones superficiales de la piel, los genitales o los ojos, siempre que la infección esté relativamente localizada; la penetración de fármacos a través de la piel se puede mejorar mezclándolos con sustancias como el polietilenglicol. La administración parenteral es la única opción en el caso de algunos fármacos y puede, en todo caso, ser necesaria para infecciones sistémicas graves; la infusión intravenosa generalmente requiere hospitalización. Algunos medicamentos tienen que usarse en concentraciones muy altas y potencialmente tóxicas debido a su poca solubilidad o poca penetración en las células. A veces se puede lograr el suministro de concentraciones antivirales de compuestos a las células incorporando el fármaco en liposomas o conjugando el compuesto a un ancla de membrana hidrofóbica. También puede ser necesaria una química sofisticada para modificar los antivirales potenciales, como los péptidos u oligonucleótidos sintéticos, que de otro modo se degradarían rápidamente intra o extracelularmente. Algunos medicamentos antivirales experimentales se conjugan con anticuerpos antivirales, o se incorporan en liposomas recubiertos con dicho anticuerpo, para dirigirlos a las células infectadas por virus. Aparición de mutantes resistentes a los medicamentos Con casi todos los agentes antivirales nuevos, pronto surgen mutantes resistentes a los medicamentos in vitro e in vivo, especialmente durante la terapia a largo plazo de infecciones crónicas y entre pacientes inmunocomprometidos (Cuadro 4.5). En su forma más simple, la resistencia puede deberse a una mutación de un solo punto en el gen que codifica la proteína viral particular que es el objetivo del compuesto. Pueden ocurrir aumentos escalonados en el grado de resistencia a medida que se acumulan más sustituciones de nucleótidos, a menudo en un orden particular. Las cepas resistentes a los medicamentos a menudo muestran menos capacidad de replicación y/o menos virulencia, y pueden ser reemplazadas por virus de tipo salvaje en un cultivo celular o en un paciente, a menos que se mantengan por la presión de selección de la presencia continua del fármaco. Sin embargo, como demostración del poder de selección, tales cepas resistentes también pueden desarrollar mutaciones compensatorias adicionales que mejoran la capacidad de replicación. Los aislamientos de virus clínicos pueden analizarse para determinar la sensibilidad a los medicamentos mediante el crecimiento en células cultivadas en presencia de diluciones en serie del agente (un ensayo "fenotípico").

Para las combinaciones de fármacos en las que la resistencia se asocia regularmente con mutaciones particulares, la secuencia del genoma del aislado del paciente se compara con una biblioteca de secuencias conocidas; esto permite la predicción del patrón de resistencia del nuevo aislado y la elección de la terapia antiviral óptima. Este enfoque está más desarrollado en el caso del VIH. Por supuesto, a menudo la primera indicación durante el tratamiento a largo plazo de una infección persistente de que se ha desarrollado resistencia puede ser clínica, es decir, un deterioro de los síntomas del paciente o un aumento de la carga viral. Las mutaciones de resistencia solo pueden desarrollarse mientras un virus se está replicando, una situación que es más probable que se desarrolle durante la monoterapia donde la concentración del fármaco es intermedia, es decir, suficiente para crear una presión de selección, pero no lo suficientemente alta como para evitar la replicación por completo.
Los principios necesarios para minimizar este problema se comprenden bien a partir de la experiencia con la resistencia a los antibióticos en bacterias y la quimioterapia contra el cáncer. Los agentes antivirales deben administrarse en dosis suficientemente altas y sin interrupción, de modo que se mantengan los niveles del fármaco inhibidor. La terapia de combinación con dos o más agentes (preferiblemente con distintos modos de acción) es teóricamente atractiva y se ha demostrado que con el VIH retrasa en gran medida la aparición de resistencia y mejora la respuesta terapéutica. Además, si esto permite que uno o ambos medicamentos se administren en dosis más bajas, la terapia combinada puede reducir la incidencia de efectos secundarios tóxicos.
4.4 Estrategias clínicas
Los medicamentos antivirales se usan con éxito teniendo en cuenta varias estrategias diferentes, que incluyen:
1. Terapia a largo plazo en infecciones crónicas. Es importante darse cuenta de que se pueden abordar diferentes objetivos terapéuticos posibles, por ejemplo (a) erradicación del virus del cuerpo, (b) supresión de la replicación del virus para que la carga viral en el plasma se reduzca en gran medida o sea indetectable, (c) retrasar o prevenir la enfermedad clínica, la progresión de la enfermedad o la muerte, o (d) mejorar los marcadores de laboratorio de la enfermedad, por ejemplo, pruebas de función hepática (hepatitis) y recuento de células CD4 (VIH). Estas opciones deben entenderse claramente tanto en la planificación del tratamiento clínico como en el diseño de ensayos de fármacos. El resultado de un curso de tratamiento a largo plazo se designa como IR (respuesta inicial), ETR (respuesta al final del tratamiento, es decir, posiblemente seguida de rebote) o SR (respuesta sostenida, es decir, sin rebote).
2. Tratamiento inmediato de la infección aguda, por ejemplo, inhibidores de la neuraminidasa para la gripe, aciclovir o un derivado para la infección aguda por herpes simple. Como en muchos de los casos, en estas infecciones, la replicación del virus alcanza su punto máximo al principio de la enfermedad clínica y disminuye posteriormente: se ha demostrado que un inicio rápido del tratamiento es muy importante para lograr un beneficio óptimo.
3. Profilaxis contra la progresión o el recrudecimiento de la enfermedad, o para prevenir la diseminación viral. Pacientes que sufren de recrudecimiento frecuente de los genitales el herpes puede transformar sus vidas con una terapia de mantenimiento diario a largo plazo con aciclovir, famciclovir o valaciclovir. De manera similar, el uso de valganciclovir en pacientes trasplantados, ya sea profilácticamente para todos aquellos con anticuerpos CMV preexistentes (es decir, infección persistente), o de forma preventiva para aquellos que tienen evidencia de laboratorio de replicación de CMV, ha llevado a una reducción importante en CMV. enfermedad entre estos pacientes.
4.5 Prevención de la transmisión de virus
Sin intervención, aproximadamente el 25% de los bebés nacidos de madres infectadas por el VIH se infectan. Esto se puede reducir a aproximadamente el 2%, un resultado dramáticamente importante, mediante una combinación apropiada de tres medicamentos antivirales administrados a la madre al final del embarazo y al bebé después del nacimiento, combinado con una cesárea si no se ha logrado la supresión viral para el momento. del trabajo de parto, y combinado con la evitación de la lactancia materna. Otros regímenes incluyen zidovudina intravenosa o nevirapina como un curso corto o una dosis única para la madre y el bebé en el momento del parto. Los regímenes de medicamentos antivirales también se usan para la protección posterior a la exposición contra la transmisión del VIH a individuos después de una exposición sexual conocida al VIH. Recientemente, se aprobó la profilaxis diaria previa a la exposición para personas que se cree que tienen un alto riesgo continuo de contraer el VIH, utilizando la tableta combinada Truvada (tenofovir y emtricitabina). Una adopción más amplia de este enfoque en el futuro podría incluso generar cierta similitud entre la prevención del VIH y la profilaxis de la malaria.
4.6 Mecanismo de acción y papel de los antivirales individualmente
4.6.1 Interferones
Los interferones (IFN) han ofrecido ventajas potenciales como agentes antivirales ideales desde que se descubrieron en 1957. Estos son productos celulares naturales de la infección viral y muestran un amplio espectro de actividad contra prácticamente todos los virus Los primeros ensayos clínicos se realizaron con cantidades inadecuadas de interferones semipurificados producidos mediante el tratamiento de leucocitos o fibroblastos humanos cultivados con un paramixovirus o con un ARN sintético de doble cadena. Sin embargo, la clonación y expresión del gen del interferón α humano (IFN-α) en Escherichia coli en 1980 permitió producir cantidades terapéuticas de IFN-α a un costo muy reducido, lo que permitió explorar su verdadero papel clínico. Desde entonces, los genes para todos los subtipos conocidos de IFN-α humano, así como IFN-β e IFN-γ, se han clonado en células procariotas y/o eucariotas. Los interferones no son efectivos por vía oral y, por lo tanto, deben administrarse mediante inyección. El IFN-α es mucho más activo in vivo que el IFN-β o el IFN-γ, probablemente porque estos últimos no alcanzan ni mantienen los niveles sanguíneos requeridos después de la administración intramuscular. Los efectos secundarios tóxicos se observan regularmente y pueden ser significativos con dosis superiores a 107 unidades por día, incluso cuando se emplean subtipos de IFN clonados altamente purificados. La fiebre suele acompañar a las dosis altas, pero dura solo un día más o menos. La fatiga intensa es el síntoma más debilitante y puede estar acompañada de malestar general, anorexia, mialgia, dolor de cabeza, náuseas, vómitos, pérdida de peso, eritema y sensibilidad en el lugar de la inyección, alopecia parcial (reversible), sequedad de boca, neuropatía sensorial periférica reversible, o signos atribuibles al sistema nervioso central. La depresión es un efecto secundario común ya veces grave. Si se prolonga la administración de dosis altas de interferón, se observan regularmente varios indicadores de mielosupresión (granulocitopenia, trombocitopenia y leucopenia) y pruebas de función hepática anormales, ambas reversibles al suspender el tratamiento. A pesar de su promesa, solo hay un número limitado de situaciones en las que el interferón-α se ha convertido en la terapia estándar. Estos incluyen hepatitis C (en combinación con ribavirina), hepatitis B y sarcoma de Kaposi relacionado con el SIDA. Las verrugas genitales se han tratado con éxito, y la papilomatosis laríngea juvenil, una afección grave que requiere extirpación quirúrgica repetida después de las recurrencias, se puede detener con una inyección local de interferón; sin embargo, los tumores reaparecen cuando se retira la terapia. Otras afecciones no virales que pueden sufrir una remisión temporal o una regresión parcial con una terapia vigorosa con interferón incluyen la leucemia de células pilosas, el melanoma maligno, la leucemia mielocítica crónica y, en el caso de la esclerosis múltiple, el interferón-β. En estas situaciones, los interferones pueden estar actuando no como antivirales sino como citocinas que ejercen efectos inmunomoduladores. Debido al patrón de efectos secundarios anterior, el tratamiento sistémico a largo plazo con interferón a menudo es incómodo o desagradable, y no es raro que los pacientes que se someten a un tratamiento a largo plazo para la hepatitis C abandonen su ciclo de tratamiento. El desarrollo reciente de regímenes antivirales sin interferón muy efectivos para el tratamiento de la hepatitis C es un gran avance que ha sido aclamado tanto por pacientes como por médicos.
4.6.2 Bloqueo de la adhesión o fusión
Una opción teórica para la terapia antiviral es inhibir el primer paso en el ciclo de replicación viral, a saber, la unión del virión a su receptor específico en la membrana de la célula huésped. Las sustancias diseñadas para imitar al receptor celular o al ligando viral teóricamente deberían lograr esto. Por ejemplo, la unión del VIH a su receptor (CD4) puede ser bloqueada por CD4 soluble (que se une al virión) o por un péptido sintético correspondiente al ligando de la glicoproteína gp120 de la cubierta del VIH que se une al receptor celular. Otro ejemplo de esto último es el bloqueo del correceptor de VIH CCR-5, utilizando un mimético de ligando o un anticuerpo que se une al sitio. Maraviroc es un antagonista del correceptor CCR5 ahora aprobado para el tratamiento del VIH; cuando se administra junto con el tratamiento estándar, se ha demostrado que conduce a un mejor resultado. Un problema importante con los imitadores de ligandos es que puede producirse la saturación de los receptores celulares y, por lo tanto, interferir con la función fisiológica normal de esa glicoproteína de membrana. Se ha informado que maraviroc causa reacciones alérgicas y hepatotoxicidad. Los imitadores de receptores pueden ser seguros, pero sería necesario confirmar que no provoquen una respuesta autoinmune. La cristalografía de rayos X ha proporcionado información detallada sobre el sitio de unión de una amplia gama de agentes antivirales que bloquean la eliminación de la cubierta de los picornavirus. Muchos de los estudios realizados hasta la fecha han utilizado el rinovirus humano tipo 14 (HRV-14) como modelo. La mayoría de las drogas, a pesar de la diversidad en la estructura química, se unen al mismo sitio en HRV-14, a saber, un bolsillo hidrofóbico que se encuentra inmediatamente debajo del suelo del cañón que comprende el ligando (sitio de unión al receptor) en la proteína de la cápside viral VP1. Después de la unión del fármaco, las interacciones hidrofóbicas dan como resultado la deformación del suelo del cañón; esto inhibe la unión del virión a los receptores celulares, pero, lo que es más importante, también inhibe la eliminación del revestimiento del virión. Se cree que esto ocurre al bloquear VP1 en una conformación, evitando así el desmontaje del virión que normalmente ocurre en el entorno ácido del endosoma. Cuando se administran de forma profiláctica en lugar de terapéutica, se ha afirmado que los antivirales de esta naturaleza reducen los síntomas de los resfriados comunes inducidos por ciertos serotipos de rinovirus, pero no por otros.
4.6.3 Bloqueo del revestimiento: bloqueadores de canales iónicos
En la década de 1960, se sintetizó amantadina, una amina simétrica simple de tres anillos, y se demostró que inhibía la replicación de los virus de influenza A (pero no de los virus de influenza B). El objetivo principal de la amantadina es la proteína M2, que es un componente menor de la envoltura viral de la influenza. La influenza B carece de una proteína M2 y, por lo tanto, se puede entender la especificidad de la amantadina para la influenza A. M2 forma un canal iónico transmembrana tetramérico que reduce el gradiente de pH a través de la envoltura de los viriones entrantes dentro de los endosomas ácidos. También se cree que juega un papel en ayudar al transporte de HA recién sintetizado a la membrana plasmática al reducir los gradientes transmembrana a través de las cisternas trans-Golgi. Por lo tanto, la amantadina actúa en estos dos pasos distintos del ciclo de replicación como bloqueador de los canales iónicos. En primer lugar, al elevar el pH del endosoma, la amantadina previene el cambio conformacional mediado por el pH 5 en la molécula de HA necesaria para la fusión de la envoltura viral con la membrana endosómica. En segundo lugar, más adelante en el ciclo de replicación, al alterar el entorno iónico dentro de la vía exocítica, la amantadina evita que la HA recién sintetizada asuma la conformación correcta para incorporarse a la envoltura de los viriones en ciernes. Terapéuticamente, se ha informado que la amantadina reduce la gravedad de los síntomas en aproximadamente el 50% de los casos, pero solo si se administra dentro de las primeras 24 a 48 horas y en dosis altas. Administrado profilácticamente, puede reducir significativamente la incidencia de influenza clínica (50% a 90% en varios ensayos). Sin embargo, en la práctica, su uso como profiláctico exige la ingesta de 200 mg diarios durante 1 a 2 meses desde el inicio de una epidemia de influenza en la comunidad, y es claro que la vacunación presenta una alternativa más segura y económica. La amantadina tiene una ventana terapéutica estrecha, con efectos secundarios que ocurren comúnmente en dosis que no exceden mucho la dosis terapéutica. Dichos efectos secundarios se relacionan principalmente con el sistema nervioso central (pérdida de concentración, insomnio, nerviosismo, mareos, somnolencia, ansiedad, confusión), pero generalmente son reversibles y leves. En 1969 se descubrió que el fármaco aliviaba los síntomas de la enfermedad de Parkinson y otros síndromes extrapiramidales, y en la actualidad se usa más para esta indicación que como antiviral. La rimantadina (fig. 4.4), es un derivado metilado que presenta menos efectos secundarios sobre el sistema nervioso central y es el fármaco de elección en la mayoría de los casos. Ambos fármacos se administran por vía oral, pero cualquiera de ellos puede administrarse a pacientes hospitalizados mediante un aerosol. Como la mayoría de los aislamientos recientes de H3N2 y H1N1 pandémico muestran resistencia a los adamantanos, los nuevos inhibidores de la neuraminidasa han reemplazado en gran medida el uso de ambos medicamentos.
Aunque los compuestos anteriores son relativamente específicos para el virus de la influenza A, diferentes proteínas transmembrana hidrofóbicas cortas que funcionan como canales de iones son codificado por varios virus diferentes; estos incluyen la proteína vpu del VIH, la proteína E del SARS-CoV y la proteína p7 del VHC. Los fármacos que actúan como bloqueadores de los canales iónicos contra algunos de estos están en desarrollo activo y pueden representar el prototipo de una nueva clase de compuestos activos contra muchos otros virus. Inhibidores de la ADN polimerasa viral. Muchos de los inhibidores exitosos de la replicación viral son análogos de nucleósidos, con actividad antiviral particularmente contra los herpesvirus o el VIH. Los primeros prototipos, como el arabinósido de adenina, son inhibidores relativamente indiscriminados de la síntesis de ADN celular y viral. Es comprensible que estos primeros compuestos a menudo produjeran efectos secundarios tóxicos, dirigidos especialmente a las células en división en la médula ósea y el tracto gastrointestinal. Acicloguanosina (Aciclovir) y homólogos Un gran avance en la quimioterapia antiviral ocurrió en 1977 cuando Elion y sus colegas desarrollaron un profármaco que depende de una enzima viral para convertirlo en su forma activa. La acicloguanosina, ahora comúnmente conocida como aciclovir, es un derivado de la guanina con una cadena lateral acíclica, cuyo nombre químico completo es 9-(2-hidroxietoximetil) guanina (fig. 4.1). Su ventaja única sobre los derivados de nucleósidos anteriores es que la enzima codificada por herpesvirus, la timidina cinasa (TK), que tiene una especificidad más amplia que la TK celular, es necesaria para fosforilar intracelularmente la acicloguanosina a monofosfato de acicloguanosina (ACG-P); una cinasa GMP celular luego completa la fosforilación al principio activo, trifosfato de acicloguanosina (ACG-PPP) (fig. 4.2). Además, ACG-PPP inhibe la ADN polimerasa codificada por herpesvirus en menos 10 veces más eficaz que la ADN polimerasa celular α. Actúa como inhibidor y sustrato de la enzima viral, compitiendo con GTP e incorporándose al ADN: el resultado es la terminación de la cadena, ya que el aciclovir carece del grupo 3'-hidroxilo necesario para la elongación de la cadena. Dado que la activación del profármaco necesita la TK viral, el aciclovir es esencialmente no tóxico para las células no infectadas, pero es un poderoso inhibidor de la síntesis de ADN viral en las células infectadas, lo que le otorga una selectividad mucho mayor que en el caso de los análogos de nucleósidos anteriores. Los virus del herpes simple tipo 1 y 2 (HSV-1 y -2) son muy sensibles al aciclovir; El virus varicela-zoster (VZV) es susceptible a concentraciones algo más altas del fármaco.
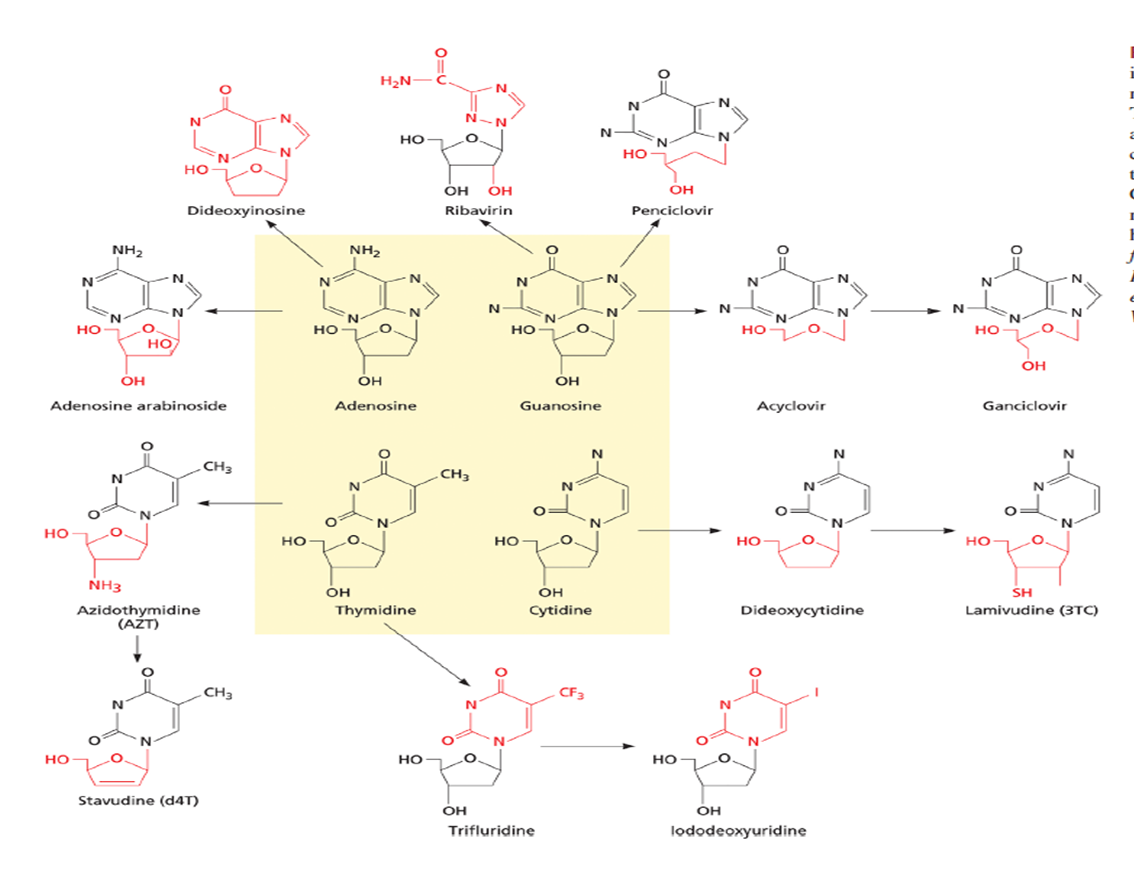
FIGURA 4.1. Los inhibidores de la replicación viral son análogos de nucleósidos o nucleótidos. Los cuatro desoxinucleótidos naturales están resaltados en amarillo caja central, y las flechas conectan estos a medicamentos antivirales relacionados. Modificaciones químicas dando aumento de cada fármaco antiviral son resaltado en rojo.
La sensibilidad relativa de los diferentes virus del herpes parece depender de una interacción bastante compleja de al menos tres variables: (1) la eficiencia de la TK codificada por el virus (si la hay) para convertir el aciclovir en ACG-P, (2) la eficiencia de la TK celular quinasas en la conversión de este intermedio a ACG-PPP, y (3) la susceptibilidad de la ADN polimerasa viral a ACG-PPP. El aciclovir (Zovirax) se puede administrar por vía oral, por infusión intravenosa lenta o por vía tópica como una crema acuosa. Como se anticipó a partir de los estudios in vitro, el fármaco es esencialmente no tóxico. Los niveles de aciclovir deben controlarse cuidadosamente en pacientes con deshidratación o insuficiencia renal, ya que el fármaco, que se excreta sin cambios a través de los riñones, es relativamente insoluble y puede ocurrir cristaluria. Sin embargo, su uso ha revolucionado el tratamiento y la supresión de las infecciones mucocutáneas graves por herpes simple, la encefalitis por herpes simple y las infecciones diseminadas por HSV, varicela-zoster y CMV.
El aciclovir es un profármaco inactivo y debe ser fosforilado al compuesto trifosfato activo dentro de la célula. El primer paso, hacia el monofosfato, lo lleva a cabo la timidina quinasa viral pero no las quinasas de la célula huésped, por lo que este paso solo puede ocurrir en células infectadas. El monofosfato luego se convierte en el trifosfato activo mediante quinasas celulares. ACV-TTP luego inhibe la ADN polimerasa viral 10 veces más que la ADN polimerasa α del huésped; además, cuando se incorpora al ADN en crecimiento, actúa como un terminador de cadena porque no tiene grupo hidroxilo 3 'del anillo de azúcar.
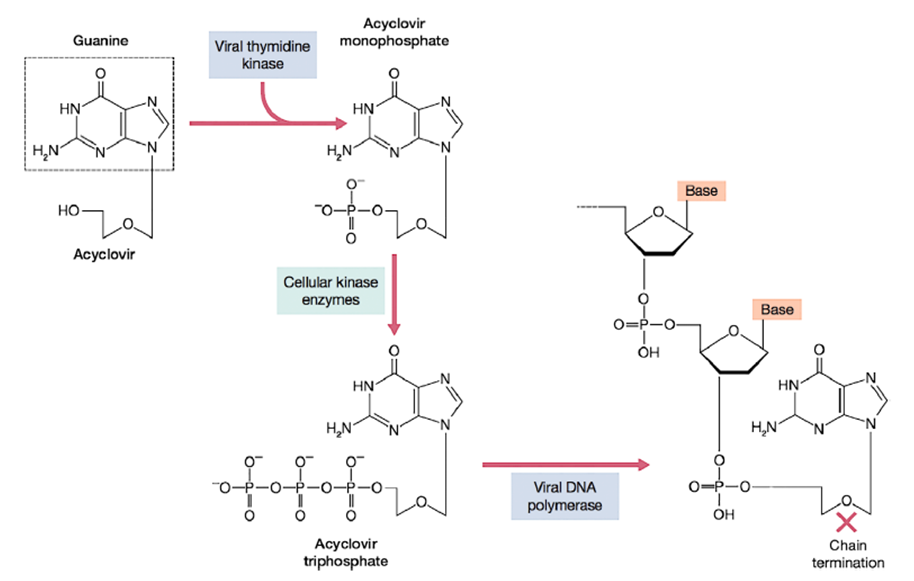
FIGURA 4.2 Mecanismo de inhibición selectiva de la replicación del herpesvirus por aciclovir.
El aciclovir tiene las limitaciones de una baja absorción oral y una vida media corta, por lo que normalmente debe tomarse cinco veces al día. Los derivados más recientes del aciclovir son el valaciclovir (un éster de valina del aciclovir), que se absorbe de tres a cinco veces mejor que el aciclovir después de la administración oral, y el famciclovir, que también se absorbe bien y se metaboliza rápidamente al compuesto activo penciclovir. Estos dos últimos fármacos tienen vidas medias más prolongadas y requieren dosis menos frecuentes que el aciclovir. Como era de esperar, los mutantes TK−resistentes a aciclovir tienen una resistencia cruzada con análogos de nucleósidos relacionados, pero no con foscarnet o el análogo de nucleótido cidofovir, que por lo tanto pueden usarse para tratar pacientes infectados (fig. 4.3).
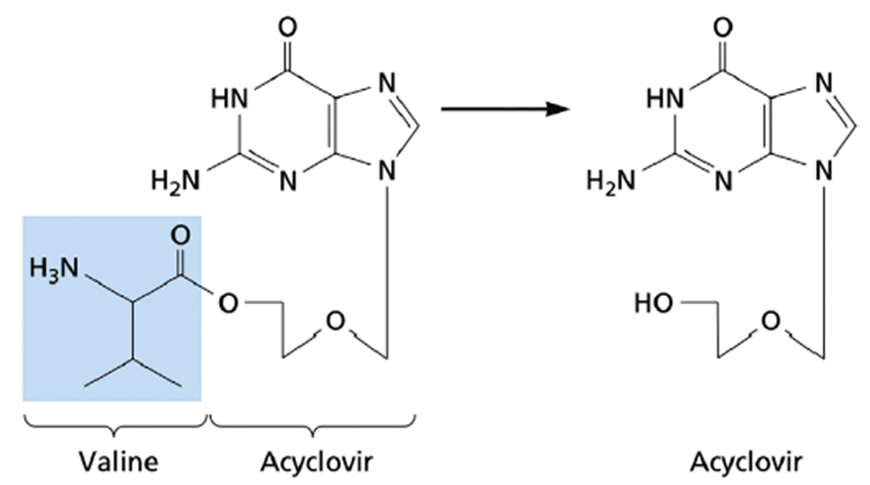
FIGURA 4.3 Estructura de valaciclovir. El valaciclovir, el éster de valina del aciclovir, se absorbe del intestino a la circulación de tres a cinco veces más rápido que el aciclovir. Una vez absorbidas por la célula, las enzimas del huésped se separan de la cadena lateral de valina. Su vida media más prolongada en el cuerpo significa que las dosis no necesitan ser tan frecuentes como las del compuesto original aciclovir.
4.6.3 Inhibidores de la replicación de virus de ARN
Un análogo de nucleósido bastante inusual, 1-β-d-ribofuranosilo, 2,4-triazol-3-carboxamida, conocido como ribavirina (fig. 4.1), se sintetizó por primera vez en 1972. A pesar de que inhibe el crecimiento de un amplio espectro de virus de ARN y ADN en células cultivadas y animales de experimentación, todavía es solo aprobado en la mayoría de los países para una gama limitada de indicaciones. Estas son infecciones crónicas por VHC en combinación con interferón (aunque este papel ahora ha sido reemplazado en gran medida por el uso de medicamentos antivirales de acción directa); e infecciones graves por el virus respiratorio sincitial, en las que se administra mediante un nebulizador para generar un aerosol de partículas pequeñas que se administra a través de una máscara o tienda de oxígeno durante 3 a 6 días. Cuando se administra por vía oral a la dosis habitual de aproximadamente 1 gramo por día, una minoría sustancial de los receptores desarrolla una anemia reversible con un mayor número de reticulocitos y niveles elevados de bilirrubina sérica: se han demostrado efectos inmunosupresores y teratogénicos en animales. Su mecanismo de acción no está claro. El hecho de que el monofosfato de ribavirina inhiba la enzima celular IMP deshidrogenasa, disminuyendo el conjunto de GTP, así como inhibiendo la protección 5′ del ARNm mediada por guanililtransferasa, sugiere que puede estar actuando en vías celulares que son algo más críticas para el virus que para el virus. célula. También se ha propuesto que puede inhibir las ARN polimerasas virales y/o aumentar la tasa de mutación de la polimerasa viral a un nivel "catastrófico" en el que se producen pocos genomas funcionales. El derivado de la 3-carboxamidina viramidina (taribavirina), un profármaco de la ribavirina, tiene un espectro de actividad similar pero puede tener una toxicidad ligeramente menor. La ribavirina oral o intravenosa se ha utilizado en las fiebres hemorrágicas virales, incluidas las fiebres hemorrágicas de Lassa, Crimea-Congo y la infección por Hantavirus, y existe cierta evidencia de beneficio. Inhibidores del nucleósido de la transcriptasa inversa.
4.6.4 Inhibidores de la transcriptasa inversa (RT)
La llegada del SIDA a principios de la década de 1980 estimuló una importante búsqueda de nuevos enfoques para la terapia antiviral. Pocos en la comunidad médica y científica tenían experiencia con lentivirus y no había agentes antirretrovirales efectivos. A partir de entonces se lograron grandes avances rápidamente y nació una nueva era de quimioterapia viral. El primer compuesto que mostró suficiente actividad antiviral in vivo para ser autorizado para uso humano fue la 3′-azido-2′,3′-didesoxitimidina, también conocida como azidotimidina, AZT o zidovudina (Fig. 12.1), un inhibidor de la transcriptasa inversa. A diferencia del aciclovir, el AZT es fosforilado por las quinasas celulares a AZT trifosfato (AZT-PPP), que ejerce su efecto antiviral contra el VIH al inhibir la transcriptasa inversa del VIH, siendo aceptado por la enzima con preferencia a la TTP. AZT-PPP se une a la transcriptasa inversa aproximadamente 100 veces más eficientemente que a la ADN polimerasa celular α. AZT-PPP se incorpora a la cadena de ADN del VIH en crecimiento, lo que lleva a la terminación prematura de la cadena. Dado que la actividad de AZT no depende de la fosforilación por enzimas virales, es menos selectivo y más tóxico que el aciclovir. Claramente, el AZT suprime la replicación pero no elimina el ADN proviral de las células infectadas. El objetivo final de la erradicación del VIH del cuerpo solo puede lograrse si se previene el reclutamiento de la infección a nuevas células y si las células ya infectadas (el “reservorio” del VIH) eventualmente mueren. La toxicidad del AZT es un problema grave, que incluye supresión de la médula (neutropenia, anemia macrocítica), atrofia reversible de los músculos proximales, náuseas y dolores de cabeza.
4.6.5 Inhibidores de la transcriptasa inversa no nucleósidos
A fines de la década de 1980, se desarrollaron varias clases de inhibidores ("TIBO" y "HEPT") y se descubrió que eran inhibidores altamente específicos y potentes de la transcriptasa inversa del VIH-1. Estos fármacos se unen de forma no competitiva a un sitio hidrofóbico a 10 Å de distancia del sitio catalítico de la enzima, lo que provoca cambios conformacionales en el dominio del pulgar p66 de la transcriptasa inversa, lo que afecta la función del sitio catalítico de la transcriptasa inversa. El primer inhibidor de la transcriptasa inversa no nucleósido que se aprobó (en 1997) fue la nevirapina, seguida de la delavirdina y el efavirenz, mientras que la etravirina (aprobada por la Agencia de Alimentos y Medicamentos de los Estados Unidos en 2008) tiene una estructura ligeramente diferente y es valiosa en pacientes con virus resistentes a los inhibidores de la transcriptasa inversa no nucleósidos anteriores. Estos se absorben bien y la vida media prolongada asociada en el cuerpo significa que la dosis de una o dos veces al día es efectiva. Los efectos secundarios incluyen hepatotoxicidad y manifestaciones psiquiátricas. Dado que los inhibidores de la transcriptasa inversa no nucleósidos tienen un objetivo molecular y un mecanismo de acción diferentes de los inhibidores de la transcriptasa inversa no nucleósidos, estos son valiosos en combinación con los inhibidores de la transcriptasa inversa no nucleósidos para retrasar la aparición de resistencia a los medicamentos y, de hecho, los dos juntos a lo largo de wiLos inhibidores de la proteasa son un pilar de la terapia actual. La aparición de resistencia a todos los fármacos anteriores es un problema continuo que requiere un manejo cuidadoso. El entecavir es un análogo de nucleósido introducido más recientemente que es particularmente activo contra la ADN polimerasa del virus de la hepatitis B, pero no contra la transcriptasa inversa del VIH.
4.6.6 Inhibidores de proteasas virales
La escisión de proteínas virales por proteasas es esencial en varias etapas de muchos ciclos de replicación viral: los ejemplos incluyen la activación de glicoproteínas de fusión de la envoltura, la activación de algunas enzimas virales, la escisión postraduccional del producto poliproteico del ARNm y la maduración del virión. Como muchas de las proteasas están codificadas por virus, debería ser posible encontrar agentes que inhiban específicamente una proteasa viral sin interferir con las proteasas celulares esenciales. El desarrollo de inhibidores de la proteasa del VIH es el primer ejemplo de este enfoque. La estructura tridimensional de la proteasa del VIH se resolvió mediante cristalografía de rayos X. Se identificó el sitio activo y se encontró que albergaba siete aminoácidos, utilizando enzimas producidas en grandes cantidades mediante tecnología de ADN recombinante. Se descubrió que la enzima escindía secuencias que contenían los dipéptidos Tyr-Pro o Phe-Pro (fig. 4.4).
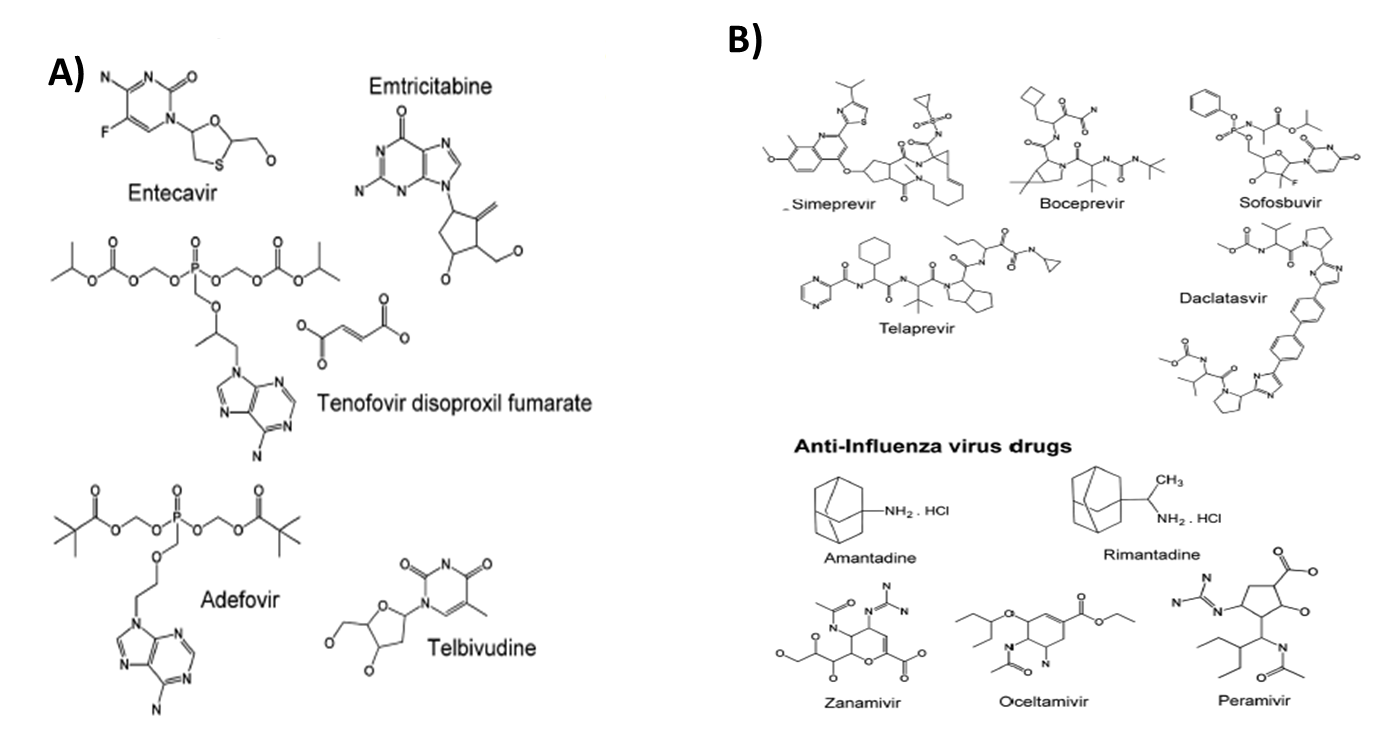
FIGURA 12.4 Estructuras químicas de algunos agentes antivirales utilizados contra la hepatitis B, hepatitis C y virus de la influenza. (A) compra de anti-VHB; (B) Anti-HCV e Influenza comprados.
Cuando un enlace de hidroxietileno reemplaza el enlace peptídico, no se produce la escisión; Se siguen produciendo proteínas virales, pero las partículas virales que brotan de las células son inmaduras y no infecciosas. Una variedad de diferentes inhibidores que se unen y bloquean el sitio activo ahora se usan de forma rutinaria, generalmente como parte de una terapia combinada (Tabla 4.1; Fig. 4.5). Como era de esperar, pueden surgir cepas de VIH resistentes, muchas de las cuales son exclusivas de un inhibidor en particular. Los efectos secundarios, que pueden ser molestos, incluyen náuseas, vómitos, diarrea, hiperlipidemia, resistencia a la insulina, redistribución de la grasa corporal a las regiones del tronco y función hepática anormal.
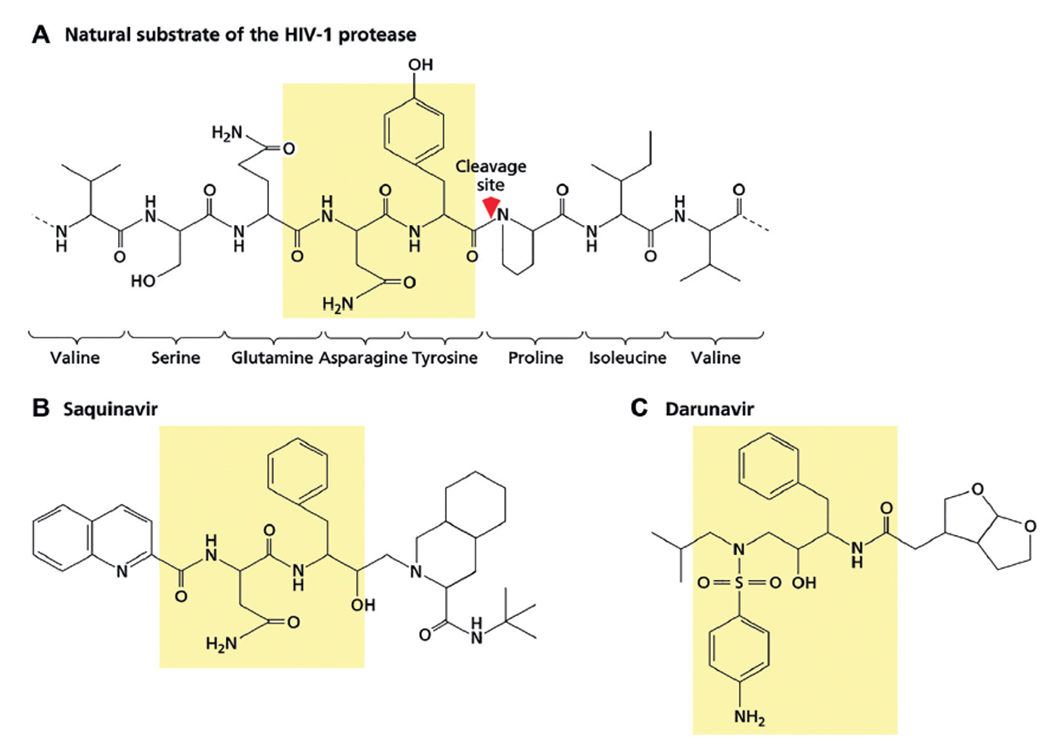
FIGURA 4.5 Comparación entre uno de los sitios cortados por la proteasa del VIH y el inhibidor de la proteasa saquinavir. (A) Secuencia de aminoácidos dentro de la proteína Gag-Pol que muestra un sitio de escisión (entre tirosina y prolina, mostrado por la flecha roja). (B y C) Las estructuras correspondientes del inhibidor de la proteasa saquinavir.
4.6.7 Inhibidores de la neuraminidasa
El desarrollo de inhibidores de la neuraminidasa altamente efectivos se volvió factible cuando el análisis de la estructura tridimensional y estructura del sitio catalítico. Dos agentes exitosos, zanamavir (administrado por vía intranasal) y oseltamivir (administrado por vía oral), se han vuelto importantes en el tratamiento y la profilaxis de la influenza. Ambos se unen fuertemente al sitio activo de la enzima y previenen la liberación de viriones de la progenie, evitando así la infección de nuevas células y la propagación adicional de la infección. Cuando se usa dentro de las 24 a 30 horas posteriores al inicio de los síntomas, cualquiera de los medicamentos puede acortar la duración de los síntomas de uno a tres días, y también son útiles como tratamiento profiláctico. En contraste con los adamantanos, el zanamavir y el oseltamivir causan muy poca toxicidad y están asociados con menos problemas de resistencia a los medicamentos. Cada uno tiene un amplio espectro de acción en las cepas de influenza tanto de tipo A como de tipo B, y una baja tasa de resistencia a los medicamentos hace que cada uno sea un componente importante en la planificación de futuras pandemias. Se mantienen reservas de medicamentos para brindar profilaxis y tratamiento rápidos, si es necesario en el período previo antes de que esté disponible una vacuna específica contra una nueva cepa pandémica. Sin embargo, el costo y la logística, por supuesto, limitarán la extensión y la duración de dicho tratamiento frente a una nueva pandemia. Los inhibidores de la neuraminidasa desarrollados más recientemente incluyen peramivir, un agente intravenoso autorizado para uso de emergencia en pacientes hospitalizados que sufren de influenza H1N1 2009, y laninamivir, que es activo contra los virus resistentes al oseltamivir y tiene un efecto tan duradero que una sola inhalación en el primer día de tratamiento parece ser eficaz.